Frataksin je protein koji je kod ljudi kodiran genom FXN.[5][6]
Kratke činjenice Dostupne strukture, PDB ...
| FXN |
|---|
 |
| Dostupne strukture |
|---|
| PDB | Pretraga ortologa: PDBe RCSB |
|---|
| Spisak PDB ID kodova |
|---|
1EKG, 1LY7, 3S4M, 3S5D, 3S5E, 3S5F, 3T3J, 3T3K, 3T3L, 3T3T, 3T3X |
|
|
| Identifikatori |
|---|
| Aliasi | FXN |
|---|
| Vanjski ID-jevi | OMIM: 606829 MGI: 1096879 HomoloGene: 47908 GeneCards: FXN |
|---|
|
|
|
|
| Ortolozi |
|---|
| Vrste | Čovjek | Miš |
|---|
| Entrez | | |
|---|
| Ensembl | | |
|---|
| UniProt | | |
|---|
| RefSeq (mRNK) | | |
|---|
| RefSeq (bjelančevina) | | |
|---|
| Lokacija (UCSC) | Chr 9: 69.04 – 69.08 Mb | Chr 19: 24.24 – 24.26 Mb |
|---|
| PubMed pretraga | [3] | [4] |
|---|
| Wikipodaci |
|
Zatvori
Nalazi se u mitohondrijama, a iRNK frataksina se uglavnom eksprimira u tkivima s visokom brzinom metabolizma. Funkcija frataksina nije jasna, ali je uključen u sastavljanje klastera gvožđa i sumpora. Predloženo je da djeluje ili kao gvožđev šaperon ili kao protein za skladištenje gvožđa. Smanjena ekspresija frataksina je uzrok Friedreichove ataksije.
Dužina polipeptidnog lanca je 210 aminokiselina, а molekulska težina 23.135 Da.[7]
| 10 | | 20 | | 30 | | 40 | | 50 |
|---|
| MWTLGRRAVA | | GLLASPSPAQ | | AQTLTRVPRP | | AELAPLCGRR | | GLRTDIDATC |
| TPRRASSNQR | | GLNQIWNVKK | | QSVYLMNLRK | | SGTLGHPGSL | | DETTYERLAE |
| ETLDSLAEFF | | EDLADKPYTF | | EDYDVSFGSG | | VLTVKLGGDL | | GTYVINKQTP |
| NKQIWLSSPS | | SGPKRYDWTG | | KNWVYSHDGV | | SLHELLAAEL | | TKALKTKLDL |
| SSLAYSGKDA |
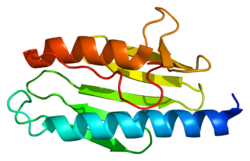
Rendgenska kristalografija pokazala je da se ljudski frataksin sastoji od β-listova koje podržava par paralelnih α-heliksa, tvoreći kompaktni αβ sendvič.[8] Frataksinski homolozi u drugih vrsta su slični, sa istom strukturom jezgra. Međutim, repne sekvence frataksina, koje se protežu od kraja jedne spirale, razlikuju se u sekvenci i razlikuju se po dužini. Ljudski frataksin ima dužu repnu sekvencu od frataksina koji se nalazi u bakterijama ili kvascima. Pretpostavlja se da je svrha repa stabilizacija proteina.[8]
Kao i većina mitohondrijskih proteina, frataksin se sintetizira u citoplazmatskim ribosomima, kao velike prekursorske molekule sa mitohondrijskim ciljanim sekvencama. Nakon ulaska u mitohondrije, molekule se proteolitskom reakcijom razgrađuju kako bi se dobio zreli frataksin.[9]
Frataksin je lokaliziran u mitohondrijama. Funkcija frataksina nije potpuno jasna, ali izgleda da je uključen u sastavljanje grupa gvožđe-sumporna grupa. Predloženo je da djeluje ili kao gvožđev šaperon ili kao protein za skladištenje gvožđa.[10]
Frataksinska iRNK pretežno je eksprimirana u tkivima sa visokom stopom metabolizma, uključujući jetru, bubrege, smeđe masti i srce. Mišji i kvaščevi frataksin homolozi sadrže potencijalnu N-terminalnu sekvencu ciljanja mitohondrija, a uočeno je da se ljudski frataksin lokalizira s proteinom mitohondrija. Nadalje, pokazalo se da poremećaj gena kvasaca dovodi do disfunkcije mitohondrija. Stoga se vjeruje da je Friedreichova ataksija mitohondrijska bolest uzrokovana mutacijom u jedarnom genomu (konkretno, ekspanzija intronskog ponavljanja tripleta GAA u genu FXN, koji kodira protein frataksin).[5][11][12]
Smanjena ekspresija frataksina je uzrok Friedreichove ataksije (FRDA), neurodegenerativne bolesti. Smanjenje ekspresije gena frataksina može se pripisati bilo utišavanju transkripcije gena za frataksin zbog epigenetičkih modifikacija u hromosomskom entitetu [13] ili od nesposobnosti prerade produženog ponavljanja GAA u prvom intronu pre-iRNK kao u bakterijama[14] i ljudskim ćelijama[15] ili oboma. Ekspanzija introna ponavljajućeg trinukleotida GAA rezultira Friedreichovom ataksijom.[16] Ovo prošireno ponavljanje uzrokuje stvaranje R-petlje, a upotreba oligonukleotida usmjerena na ponavljanje da poremeti R-petlju može reaktivirati ekspresiju frataksina.[17]
Oko 96% pacijenata sa FRDA ima ponavljajuću ekspanziju trinukleotida GAA u intronu 1 oba alela FXN gena.[18] Sve u svemu, to dovodi do smanjenja sinteze frataksinske iRNK i smanjenja (ali ne i obustavlja) sintezu proteina frataksina kod ljudi sa FRDA. (Podgrupa pacijenata sa FRDA ima ekspanziju GAA u jednom hromosomu i tačkastu mutaciju u FXN egzonu u drugom hromosomu.) U tipskom slučaju, dužina alela sa kraćom ekspanzijom GAA obrnuto korelira sa nivoima frataksina . Periferno tkivo pacijenata sa FRDA obično ima manje od 10% nivoa frataksina koje pokazuju nepogođene osobe.[18] Niži nivoi frataksina rezultiraju ranijim početkom i bržim napredovanjem bolesti.
FRDA karakteriziraju ataksija, gubitak osjetila i kardiomiopatija. Nije sasvim jasan razlog zašto nedostatak frataksina uzrokuje ove simptome. Na ćelijskoj razini, povezan je s nakupljanjem gvožga u mitohondrijama i povećanom osjetljivošću na oksidanse. Iz razloga koji nisu dobro razumljivi, ovo prvenstveno utiče na tkivo korijene dorzalnih ganglija, mali mozak i srčani mišić.[9]
Kod miševa je potpuna inaktivacija gena FXN smrtonosna u ranoj embrioskoj fazi.[19] Iako gotovo svi organizmi eksprimiraju homologe frataksina, ponavljanje GAA u intronu 1 postoji samo kod ljudi i drugih primata, pa se mutacija koja uzrokuje FDRA ne može prirodno pojaviti u drugim životinjama. Naučnici su razvili nekoliko opcija za modeliranje ove bolesti kod miševa. Jedan pristup je utišavanje ekspresije frataksina u samo jednom specifičnom tipu tkiva od interesa: srce (miševi modificirani na ovaj način nazivaju se MCK), svi neuroni (NSE) ili samo kičmena moždina i mali mozak (PRP).[20] Drugi pristup uključuje inserciju ekspanzije GAA u prvi intron mišjeg gena FXN, koji bi trebao inhibirati proizvodnju frataksina, baš kao i kod ljudi. Miševi koji su homozigotni za ovaj modificirani gen nazivaju se KIKI (skr. od eng. knock-in knock-in), a složeni heterozigoti nastali križanjem KIKI miševa s frataksinom – KIKO (skr. eng. knockout in knock-out). Međutim, čak i miševi KIKO i dalje eksprimiraju 25-36% normalne razine frataksina i pokazuju vrlo blage simptome. Konačni pristup uključuje stvaranje transgenih miševa sa GAA proširenom verzijom gena ljudskog frataksina. Ovi miševi se zovu YG22R (jedna GAA sekvenca od 190 ponavljanja) i YG22R (dvije GAA sekvence od 90 i 190 ponavljanja). Ovi miševi pokazuju simptome slične ljudskim pacijentima.[20]
Prekomjerna ekspresija frataksina u Drosophila pokazala je povećanje antioksidativnih sposobnosti, otpornost na povrede oksidativnog stresa i dugovječnost,[21] podupirući teoriju da je uloga frataksina zaštititi mitohondrije od oksidativnog stresa i nastalog ćelijskog oštećenja.
Fibroblasti iz miševskog modela fibroblasta pacijenata sa FRDA i FRDA pokazuju povećane razine dvolančanih prekida DNK.[22] Sistem isporuke gena za lentivirus je korišten za isporuku gena frataksina u FRDA model miša i ćelije ljudskih pacijenata, što je rezultiralo dugoročno obnovljenom ekspresijom frataksinske iRNK i proteina frataksina. Ova obnovljena ekspresija gena frataksina bila je praćena značajnim smanjenjem broja prekida dvostruke DNK.[22] Čini se da oštećeni frataksin u ćelijama FRDA uzrokuje smanjeni kapacitet za popravak oštećene DNK i to može doprinijeti neurodegeneraciji.[22]
Pokazano je da frataksin biološki stupa u interakciju s enzimom PMPCB.[23]
Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (Mar 1996). "Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion". Science. 271 (5254): 1423–7. Bibcode:1996Sci...271.1423C. doi:10.1126/science.271.5254.1423. PMID 8596916. S2CID 20303793. Carvajal JJ, Pook MA, dos Santos M, Doudney K, Hillermann R, Minogue S, Williamson R, Hsuan JJ, Chamberlain S (Oct 1996). "The Friedreich's ataxia gene encodes a novel phosphatidylinositol-4- phosphate 5-kinase". Nature Genetics. 14 (2): 157–62. doi:10.1038/ng1096-157. PMID 8841185. S2CID 6324358. Adinolfi S, Iannuzzi C, Prischi F, Pastore C, Iametti S, Martin SR, Bonomi F, Pastore A (Apr 2009). "Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS". Nature Structural & Molecular Biology. 16 (4): 390–6. doi:10.1038/nsmb.1579. PMID 19305405. S2CID 205522816. Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M (Oct 1996). "Clinical and genetic abnormalities in patients with Friedreich's ataxia". The New England Journal of Medicine. 335 (16): 1169–75. doi:10.1056/NEJM199610173351601. PMID 8815938. Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M (Aug 1997). "Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin". Nature Genetics. 16 (4): 345–51. doi:10.1038/ng0897-345. PMID 9241270. S2CID 5883249. Clark E, Johnson J, Dong YN, Mercado-Ayon, Warren N, Zhai M, McMillan E, Salovin A, Lin H, Lynch DR (novembar 2018). "Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease". Neuronal Signaling. 2 (4): NS20180060. doi:10.1042/NS20180060. PMC 7373238. PMID 32714592.
- Thierbach R, Drewes G, Fusser M, Voigt A, Kuhlow D, Blume U, Schulz TJ, Reiche C, Glatt H, Epe B, Steinberg P, Ristow M (Nov 2010). "The Friedreich's ataxia protein frataxin modulates DNA base excision repair in prokaryotes and mammals". The Biochemical Journal. 432 (1): 165–72. doi:10.1042/BJ20101116. PMC 2976068. PMID 20819074.
- Montermini L, Rodius F, Pianese L, Moltò MD, Cossée M, Campuzano V, Cavalcanti F, Monticelli A, Palau F, Gyapay G (Nov 1995). "The Friedreich ataxia critical region spans a 150-kb interval on chromosome 9q13". American Journal of Human Genetics. 57 (5): 1061–7. PMC 1801369. PMID 7485155.
- Bidichandani SI, Ashizawa T, Patel PI (maj 1997). "Atypical Friedreich ataxia caused by compound heterozygosity for a novel missense mutation and the GAA triplet-repeat expansion". American Journal of Human Genetics. 60 (5): 1251–6. PMC 1712428. PMID 9150176.
- Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J (Jun 1997). "Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin". Science. 276 (5319): 1709–12. doi:10.1126/science.276.5319.1709. PMID 9180083.
- Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M (Aug 1997). "Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin". Nature Genetics. 16 (4): 345–51. doi:10.1038/ng0897-345. PMID 9241270. S2CID 5883249.
- Wilson RB, Roof DM (Aug 1997). "Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue". Nature Genetics. 16 (4): 352–7. doi:10.1038/ng0897-352. PMID 9241271. S2CID 22652291.
- Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Dürr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M (Oct 1997). "Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes". Human Molecular Genetics. 6 (11): 1771–80. doi:10.1093/hmg/6.11.1771. PMID 9302253.
- Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (Oct 1997). "Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia". Nature Genetics. 17 (2): 215–7. doi:10.1038/ng1097-215. PMID 9326946. S2CID 23151137.
- Jiralerspong S, Liu Y, Montermini L, Stifani S, Pandolfo M (1997). "Frataxin shows developmentally regulated tissue-specific expression in the mouse embryo". Neurobiology of Disease. 4 (2): 103–13. doi:10.1006/nbdi.1997.0139. PMID 9331900. S2CID 6520439.
- Koutnikova H, Campuzano V, Koenig M (Sep 1998). "Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase". Human Molecular Genetics. 7 (9): 1485–9. doi:10.1093/hmg/7.9.1485. PMID 9700204.
- Zühlke C, Laccone F, Cossée M, Kohlschütter A, Koenig M, Schwinger E (Jul 1998). "Mutation of the start codon in the FRDA1 gene: linkage analysis of three pedigrees with the ATG to ATT transversion points to a unique common ancestor". Human Genetics. 103 (1): 102–5. doi:10.1007/s004390050791. PMID 9737785. S2CID 26999143.
- Bartolo C, Mendell JR, Prior TW (Oct 1998). "Identification of a missense mutation in a Friedreich's ataxia patient: implications for diagnosis and carrier studies". American Journal of Medical Genetics. 79 (5): 396–9. doi:10.1002/(SICI)1096-8628(19981012)79:5<396::AID-AJMG13>3.0.CO;2-M. PMID 9779809.
- Cossée M, Dürr A, Schmitt M, Dahl N, Trouillas P, Allinson P, Kostrzewa M, Nivelon-Chevallier A, Gustavson KH, Kohlschütter A, Müller U, Mandel JL, Brice A, Koenig M, Cavalcanti F, Tammaro A, De Michele G, Filla A, Cocozza S, Labuda M, Montermini L, Poirier J, Pandolfo M (Feb 1999). "Friedreich's ataxia: point mutations and clinical presentation of compound heterozygotes". Annals of Neurology. 45 (2): 200–6. doi:10.1002/1531-8249(199902)45:2<200::AID-ANA10>3.0.CO;2-U. PMID 9989622.
- Coppola G, De Michele G, Cavalcanti F, Pianese L, Perretti A, Santoro L, Vita G, Toscano A, Amboni M, Grimaldi G, Salvatore E, Caruso G, Filla A (maj 1999). "Why do some Friedreich's ataxia patients retain tendon reflexes? A clinical, neurophysiological and molecular study". Journal of Neurology. 246 (5): 353–7. doi:10.1007/s004150050362. PMID 10399865. S2CID 7367457.
- Branda SS, Cavadini P, Adamec J, Kalousek F, Taroni F, Isaya G (Aug 1999). "Yeast and human frataxin are processed to mature form in two sequential steps by the mitochondrial processing peptidase". The Journal of Biological Chemistry. 274 (32): 22763–9. doi:10.1074/jbc.274.32.22763. PMID 10428860.
- Gordon DM, Shi Q, Dancis A, Pain D (Nov 1999). "Maturation of frataxin within mammalian and yeast mitochondria: one-step processing by matrix processing peptidase". Human Molecular Genetics. 8 (12): 2255–62. doi:10.1093/hmg/8.12.2255. PMID 10545606.
- Forrest SM, Knight M, Delatycki MB, Paris D, Williamson R, King J, Yeung L, Nassif N, Nicholson GA (Aug 1998). "The correlation of clinical phenotype in Friedreich ataxia with the site of point mutations in the FRDA gene". Neurogenetics. 1 (4): 253–7. doi:10.1007/s100480050037. PMID 10732799. S2CID 7463903.
- Al-Mahdawi S, Pook M, Chamberlain S (Jul 2000). "A novel missense mutation (L198R) in the Friedreich's ataxia gene". Human Mutation. 16 (1): 95. doi:10.1002/1098-1004(200007)16:1<95::AID-HUMU29>3.0.CO;2-E. PMID 10874325.






