トップQs
タイムライン
チャット
視点
神経病理学
神経学の分野 ウィキペディアから
Remove ads
神経病理学(しんけいびょうりがく、英: Neuropathology)とは神経学の分野における病理学である。具体的には中枢神経系(つまり脳や脊髄)、末梢神経、筋肉などの材料を顕微鏡で観察し、病理診断や病気の原因や発生機序を研究する学問である。
この記事には複数の問題があります。 |
この記事で示されている出典について、該当する記述が具体的にその文献の何ページあるいはどの章節にあるのか、特定が求められています。 |
中枢神経病理
要約
視点
中枢神経の細胞
中枢神経の細胞は神経細胞、グリア細胞、支持組織からなる。グリア細胞はアストロサイト、オリゴデンドログリア、上衣細胞、ミクログリア、マクロファージなどが知られる。アストロサイト、オリゴデンドログリア、上衣細胞をマクログリアという。支持組織には血管、軟膜、くも膜、脈絡叢、くも膜顆粒などが含まれる。神経病理学においては神経細胞の変化は超急性期を除けばグリア細胞の反応を伴うものであり、逆にグリア細胞の反応がなければ人工的な変化の可能性が疑われるため各種細胞の同定は非常に重要と考えられている。
神経細胞
神経細胞は細胞体、軸索、樹状突起から構成される。細胞体には核、ゴルジ装置、リソソーム、ミトコンドリア、リボソーム、小胞体、リポフスチン、神経細糸、微小管、神経メラニンなどが認められる。軸索や樹状突起ではこのうちミトコンドリアや神経細糸、微小管が主に認められる。光学顕微鏡で確認できるのは核、粗面小胞体の集合物、リポフスチン、束状の神経細糸、神経メラニンなどである。
神経細胞の病理変化
神経細胞脱落
神経細胞の脱落では周囲組織の組織変化を伴うことが多い。神経細胞内物質の外部への流出(メラニン色素の遊出など)、細胞外のレヴィ小体やアルツハイマー神経原繊維変化などが知られている。細胞死に続発する変性神経細胞の処置像として、マクロファージによる神経貪食像や髄鞘破壊産物を貪食する脂肪顆粒細胞が観察される。またアストロサイトが反応しグリオーシスが形成される。
神経細胞体の形態変化
- 中心性虎斑融解
虎斑状に見えるニッスル小体(粗面小胞体)が崩壊する状態をクロマトライシスまたは虎斑融解という核周囲の細胞質の中心部が腫大し崩壊したニッスル小体が周辺に押しやられる状態を中心性虎斑融解という。これは軸索障害による逆行性変性の結果である。
- 風船状腫脹とアクロマジア
風船状腫脹(ballooning)はペラグラ、ピック病、クロイツフェルト・ヤコブ病、大脳皮質基底核変性症、進行性核上性麻痺などで記載される。風船状腫脹をした細胞がニッスル小体を失っている場合があり、その状態をアクロマジアという。
- 代謝疾患による異常代謝産物蓄積によつ腫大性変化
代謝性疾患では異常代謝産物が細胞体に蓄積されることにより腫大性変化をきたす。
- 変性疾患による異常細胞骨格成分の蓄積による腫大性変化
変性疾患では、異常蛋白などの蓄積により細胞体が腫大することがある。アルツハイマー病の神経原線維変化やレヴィ小体などが細胞体内に形成されれば全体像が腫大することがある。ballooned neuronと表現されることもある。
- 形成異常による細胞成分の異常
限局性皮質異形成や結節性硬化症の皮質結節部など細胞成分に異型が認められる疾患では神経細胞が腫大することがある。
- 神経細胞の萎縮性変化
加齢に伴う単純萎縮が知られる。またあ急激な虚血では細胞質全体が好酸性(HE染色で赤色)となり萎縮し、虚血性変化あるいは低酸素性変化という。変性萎縮した神経細胞に石灰や鉄が沈着しミネラリゼーションという変化を起こすことがある。
神経細胞質内の生理的、病的構造物
- リポフスチン
リポフスチンは神経細胞内に蓄積される消耗性色素で加齢、変性などで増加する。
- 平野小体
加齢現象に随伴する構造物で海馬の錘体細胞層に好発する。別名は好酸性棍棒状構造物。神経細胞内、神経突起内、その周辺などで認められる。生理的加齢のみならずアルツハイマー病でも多数認められる。
- 顆粒空胞変性
高齢者やアルツハイマー病などの海馬錐体細胞層の神経細胞体に好塩基性顆粒状構造物を含んだ小空胞が認められることがある。
- アルツハイマー型神経原線維変化
神経細胞内にリン酸化タウ蛋白の不溶性の蓄積が起こり、異常線維形成として蓄積したものである。古いものは好酸性を帯びてくる。強い嗜銀性があり、GB染色で黒色に明瞭に染色される。加齢や痴呆性疾患だけではなく、亜急性硬化性全脳炎、ニーマン・ピック病C型、頭部外傷、二次性タウオパチーでも出現する。
- ピック球
ピック病の海馬などの神経細胞内に形成される嗜銀性の円形構造物である。HE染色では好酸性で膨化した球状物として認識される。尖性樹状突起側に存在することがおおい。ピク病の診断的な意義をもつ神経細胞内の球状構造物である。ピック球ほど明瞭ではないものの、膨化したような球状物で細胞質が大きく腫大した神経細胞をピック細胞という。海馬歯状回、海馬支脚などが好発部位でありリン酸化タウを含んでいる。
- ポリグルコサミン小体
グルコースポリマーで形成される小体をポリグルコサン小体という。その中にはラフォラ小体、ミオクローヌス小体、ビルショウスキー小体などが含まれ、神経細胞体、神経突起、グリア細胞内に蓄積する。ラフォラ小体はラフォラ病の指標となる構造物である。ラフォラ小体は細胞体内に蓄積することが多いが神経突起で認められることもある。
- レヴィ小体
特発性パーキンソン病や加齢老人の黒質、青班核、迷走神経背側核などの脳幹の諸神経核の神経細胞室内に見られる好酸性(HE染色で赤)の小体である。典型的なものは脳幹型レヴィ小体と言われるもので中心部に強い芯があり、その周辺部に染色性の薄い帯状の空間を伴う。メラニン顆粒に埋もれていることもある。皮質型レヴィ小体は膨化した淡い球状物である。レヴィ小体はユビキチン化されており、抗ユビキチン抗体で明瞭に検出することができる。また構成蛋白であるαシヌクレインでに対する抗体でも検出ができる。αシヌクレイノパチー以外でも認められるため、神経変性に随伴する構造物である可能性もある。
- 神経細胞内好酸性顆粒
- 好塩基性封入体
若年性筋萎縮性側索硬化症の前角細胞、黒質、大脳基底核に好塩基性の大きな封入体が報告されている。
- ブニナ小体
筋萎縮性側索硬化症(ALS)の脊髄、脳幹運動神経細胞内に出現する異常構造物であり、ALSに特異的である。好酸性の小円形構造が神経細胞内の数個数珠状に連なっている。抗シスタチンC抗体で明瞭に染色される。
- スケイン様封入体
孤発性および家族性のALSの下位運動ニューロン内に好酸性の糸状、管状構造の集合体で糸を巻きとった綛のようにみえることから、この名前がついた。HE染色で同定するのは困難であるが抗ユビキチン抗体で陽性を示す。スケイン様封入体は運動ニューロン疾患以外に、進行性核上性麻痺や大脳皮質基底核変性症、ピック病、老人脳などでも線条体で高頻度に認められる。
- 球状硝子様封入体
ALSの脊髄前角細胞にスケイン様封入体よりも線維成分が密に集合したような好酸性の球状物が観察できることがある。
- レヴィ小体様硝子様封入体
ALSの脊髄前角、脳幹の運動神経細胞に時に認められるレヴィ小体に類似する円形で淡い硝子様封入体をレヴィ小体様硝子様封入体という。高度にユビキチン化しており抗ユビキチン抗体で陽性を示す。
- 痴呆症状を伴うALSに出現する歯状回神経細胞内ユビキチン化封入体
痴呆症状を伴うALSでは海馬歯状回顆粒細胞、側頭葉皮質の神経細胞にユビキチン陽性の神経細胞内の封入体が出現する。HE染色、嗜銀染色などルーチンの染色では検出困難であり抗ユビキチン抗体で陽性を示す。
- 多系統萎縮症に出現する歯状回神経細胞内ユビキチン化封入体
グリア細胞内封入体(GCI)で有名な多系統萎縮症でも神経細胞内封入体が知られている。約30%ほどに痴呆症状を伴うALSで認められる歯状回神経細胞内ユビキチン化封入体に類似する構造物が認められる。
- 多系統萎縮症の橋核神経細胞内封入体
橋核が残る神経細胞内に比較的大きな不整形の封入体が認められることがある。抗ニューロフィラメント抗体で染色される。
- ネグリ小体
狂犬病ウイルスによる好酸性の細胞質内小封入体をネグリ小体という。ウイルス性感染症では神経細胞内や各内に封入体が認められることが多い。
核内の生理的、病理的構造物
- マリネスコ小体
核小体とほぼ同じ大きさの好酸性顆粒構造物であるが臨床的意義はない。
- 各種ウイルス封入体
ウイルス性感染症では核内封入体がしばしば認められる。
- 多系統萎縮症の神経細胞核内封入体
多系統萎縮症では大脳皮質神経細胞の核内にGB染色で短い糸屑状に染色される管状構造物が確認されることがある。GB染色のみで可視化され、変性のプロセスの存在を示していると考えられている。
- トリプレットリピート病の神経細胞核内封入体
トリプレットリピート病では神経細胞体内、核内にユビキチン陽性の封入体が観察される。ボリグルタミンに対する抗体でも染色される。
神経突起の病理変化
軸索障害に関しては以下の病理学的変化が知られている。
- 順行性変性
軸索のどこかが障害され、その後の二次性変性が軸索の方向へ進展する順行性の変性進展様式を順行性変性といい、ワーラー変性ともいう。
- 逆行性変性
軸索の障害に惹起されて起こる細胞体へ向かう変性を逆行性変性という。ダイイングバック現象(dying back phenomen)ともいう。
- 経神経細胞変性
神経細胞の変性がそれと線維連絡のある別の神経細胞の変性を惹起することを経神経細胞変性という。
- 軸索反応
軸索の障害により様々な反応性の変化が起こることを軸索反応という。軸索障害後、神経細胞体へ向かう逆行性変性が起こる。その結果、神経細胞体が腫大を起こす状態を示し中心性虎斑融解を起こすことなどが典型例である。
- 索変性
神経索が変性することである。順行性、逆行性の区別はしないことが多い。
- びまん性軸索損傷
病理学的なびまん性軸索損傷と臨床のびまん性軸索損傷は異なる。軸索は断裂し髄鞘も破壊され、様々な二次反応を示す。軸索腫大(スフェロイド)も形成され特に軸索退縮球とよぶ。
- 軸索ジストロフィー
軸索腫大(スフェロイド)の1つの特殊なタイプである。軸索の遠位末端部の腫大を特徴とする。延髄ゴル核では加齢性変化や抗てんかん薬の長期服用の副作用でも出現する。
軸索樹状突起病変の基本
- 軸索萎縮と軸索消失
軸索障害の終末像は軸索の萎縮と消失である。軸索の脱落に伴い髄鞘が崩壊することも多く、その場合は破壊性分を貪食するマクロファージ、脂肪顆粒細胞が出現する。
- 軸索腫大、スフェロイド
神経突起、軸索腫大病変の総称である。軸索の近位部から最遠位部まで形成される可能性がある。HE染色では好酸性に染色されるが、ボジアン染色など嗜銀染色では一層明瞭に可視化される。ニューロフィラメントが蓄積すると考えられる。KB染色、LFB染色も用いて有髄線維のスフェロイドか無髄線維のスフェロイドかを判定する。軸索遠位部終末に形成された場合は終末ボタンといい、頭部外傷で障害部位に軸索腫大が認められた場合は軸索退縮球、軸索ジストロフィーでおこれば異栄養性軸索と呼ばれる。
プルキンエ細胞と周辺の神経突起の変化
- トルペド
プルキンエ細胞の最も近位部の軸索に生じたスフェロイドである。
- カクタス
プルキンエ細胞の細胞体あるいは樹状突起の表面から外側に向かって突起がでているように見えるものをカクタスという。
- ヒトデ小体
プルキンエ細胞の樹状突起が分子層内で腫大する変化である。ヒトデ小体または樹状突起腫脹という。
- 歯状核のグルモース変性
小脳歯状核細胞周囲で軸索末端部で無髄線維が増加(発芽)することがある。これをグルモース変性という。HE染色では好酸性を呈する雲状の構造物が集積する。小脳遠心系変性を示しており、進行性核上性麻痺や歯状核赤核淡蒼球ルイ体萎縮症などで特徴的に認められる。
- エンプティバスケット
プルキンエ細胞の脱落を示す所見である。プルキンエ細胞が消失すると篭細胞の軸索だけが空っぽのバスケットのように見える。
神経突起内の蓄積物
- 嗜銀性グレイン
海馬、海馬傍回、側頭葉において嗜銀染色で棍棒状に染色される構造物が観察される。神経突起内に異常リン酸化タウが数珠状に蓄積することが明らかになっている。
- ニューロピルスレッド
アルツハイマー神経原線維性変化が形成される病的な場合、その周囲に糸状の嗜銀性構造物を観察することがある。HE染色では識別ができないがGB染色で明瞭に検出できる。神経突起に異常リン酸化タウが蓄積したものである。
- 老人斑
老人斑はすべて神経突起の変化であるわけではないが基質に沈着しているアミロイド成分とともに、神経突起にはリン酸化タウの蓄積が生じ軸索ジストロフィーを示す。
- 神経突起内類でんぷん小体
類でんぷん小体はアストロサイトの突起内に形成されるが、軸索内に形成されることもある。
- スフェロイド
軸索腫大のことだが蓄積物という点ではニューロフィラメント蓄積と捉えることができる。
- レヴィ関連ニューライト
レヴィ小体を形成する神経細胞周囲にユビキチン陽性のやや太く腫大した神経突起を観察することができることがある。
アストロサイト
アストロサイトはHE染色では比較的明るく、やや大きな核をもち細胞質に乏しい細胞である。細胞体には核、リソソーム、グリコーゲン顆粒、ゴルジ装置、中間径フィラメントなどに富む。中間径フィラメントの構成蛋白であるグリア線維性酸性蛋白(GFAP)やビメンチンに対する抗体で染色される。正常な場合はHE染色などルーチンの染色では突起成分は可視化されず、抗GFAP抗体を用いた免疫染色や古典的なカハール染色を行わないと突起は観察されない。組織障害に反応したアストロサイトは細胞体のみならず突起も太くなり、HE染色でも容易に観察ができるようになる。
アストロサイトのタイプ
アストロサイトは存在する場所などによって形態が異なる。
- 原形質性アストロサイト
- 線維性アストロサイト
- ベルクマングリア
ベルクマングリアとは小脳皮質プルキンエ細胞層に存在するアストロサイトの呼称である。
- ミューラー細胞
ミューラー細胞とは網膜の全層を貫く突起を有するアストロサイトの亜型である。
- 放射状グリア
発生期の神経管の内側から放射状に辺縁帯に突起を伸ばしているアストロサイトであり、神経細胞の放射状移動のガイド役になる細胞である。
アストロサイトの病理変化
- 生理的組織壊死
- 突起断裂
- アストロサイト増生(astrocytosis)
何らかの原因による組織障害でアストロサイトが増生している状態である。
- 肥胖型アストロサイト
組織障害に反応し、アストロサイトの中間系フィラメントが増生し、細胞質が腫れた状態のアストロサイトを指している。比較的急性期のアストロサイトの反応性変化である。 核縁が厚く、明るい核と好酸性の豊富な細胞質をもつアストロサイトである。クロイツフェルト・ヤコブ病や進行性多巣性白質脳症では非常に大きな肥胖性アストロサイトが認められるが、どのような組織障害でも観察できる。
- 線維型アストロサイト
細胞質には乏しいが突起内の線維成分が豊富なアストロサイトである。グリオーシスに至る前段階と考えられている。
- アメボイドグリア
脳浮腫などに随伴してアストロサイトの細胞質が腫脹しアメーバ状の形態を呈する段階を有するアストロサイトをアメポイドグリア、アメーバ様グリアという。
- エンペリポレシス
1つの細胞が他の細胞に嵌入する現象であり、脳ではアストロサイトがオリゴデンドログリアを細胞質に含む所見が観察される。
- グリオーシス(gliosis)
組織反応として線維性アストロサイトの突起が進展し、障害された組織を埋め尽くす修復、瘢痕化の状態をグリオーシスという。組織障害が慢性的に進行した場合はアストロサイトの線維性分が既存の神経線維の走行を模倣しながらグリオーシスを形成しイソモルフィックグリオーシスという。比較的急激な組織破壊の場合は既存の神経線維の走行を模倣せずにアニソモルフィックグリオーシスという。
- アルツハイマー型グリア
ウィルソン病や肝性脳症を伴う肝障害の大脳基底核で核膜、核小体が目立った大きな核をもつアストロサイトが認められアルツハイマー型グリアという。
アストロサイトへの蓄積
- 類でんぷん小体(アミロイド小体)
神経細胞だけではなくアストロサイトの特記ないにもアミロイド小体、類でんぷん小体は蓄積する。加齢性の変化で疾患特異性はない。
- ローゼンタール線維
アストロサイトの突起内に形成されるHE染色で好酸性の棍棒状の硝子様構造物である。陳旧化したグリオーシスやグリオーマなどで非特異的に認められるが。アレキサンダー病では脳全体で認められ診断的価値がある。
- グリア束
- グロテスク細胞
- 泡沫状球状物
リン酸化タウのアストロサイトへの蓄積
タウオパチーでは神経細胞だけではなくグリア細胞でもタウの蓄積が認められ、診断学的な価値がある。
- 刺様アストロサイト(thorn-shaped astrocyte)
アストロサイトの細胞体から伸びる突起にリン酸化タウが蓄積したもの。タウオパチーで非特異的に認められる。
- 房状アストロサイト(tuft-shaped astrocyte)
アストロサイトの突起のうち、細胞体に近い突起にリン酸化タウが異常蓄積したもので全体が房のようにみえるものである。進行性核上性麻痺の病理診断の根拠となるものである。
- アストロサイト斑(astricytic plaque)
アストロサイトの突起内の遠位部にリン酸化タウが異常蓄積したものであり全体として斑状、あるいは花冠状にみえるものである。大脳皮質基底核変性症の病理診断学的な指標となる構造物である。
オリゴデンドログリア
アストロサイトより小さくクロマチンに富む濃染した核を有し、細胞質はわずかである。アストロサイトと比較すると突起は少ない。神経線維束に併走するように数珠状に配列するものを束間オリゴデンドログリアという。神経細胞の周囲を取り巻く衛星細胞の多くはオリゴデンドログリアである。突起の先端部が帯状になり髄鞘を形成する。オリゴデンドログリアは1つの細胞から多数の髄鞘節が形成される(1細胞多髄鞘節)。末梢神経のシュワン細胞では1つの細胞から1つの髄鞘節が形成されるのと対照的である。細胞質には粗面小胞体、リボソーム、ミトコンドリアに富んでいる。
オリゴデンドログリアの病理変化

- 急性腫脹
オリゴデンドログリアはアストロサイトよりも脳浮腫では浮腫性変化を起こしやすい。
- 神経細胞/傍血管サテライトーシス
大脳白質の異所性神経細胞や血管周囲にオリゴデンドログリアのサテライトーシスが顕著な場合は微小形成不全の所見と考えられている。
- 核内封入体
進行性多巣性白質脳症や亜急性硬化性全脳炎では神経細胞以外にオリゴデンドログリアの核内にウイルス封入体が認められる。
- グリア細胞質内封入体(glial cytoplasmic inclusion、GCI)
多系統萎縮症のオリゴデンドログリア細胞質に形成されるオタマジャクシ様の嗜銀性封入体であり、ユビキチン陽性、αシヌクレイン陽性を呈する。
リン酸化タウ蛋白のオリゴデンドログリア内の異常蓄積
タウ蛋白の異常を伴う神経変性疾患ではオリゴデンドログリア内にも蓄積が認められ、免疫染色やガリアス染色で可視化される。
- グリアコイル状小体(glial coiled body)
タウオパチーで非特異的に認められる所見である。進行性核上性麻痺、大脳皮質基底核変性症、アルツハイマー病などでオリゴデンドログリアの核の周囲にコイル状取り巻いている像が認められる。これをグリアコイル状小体という。
- 嗜銀性スレッド(argyrophilic thread)
タウオパチーにおいてオリゴデンドログリアの細胞質ではなく、突起部分に異常リン酸化タウが沈着し、免疫染色、ガリアス染色などで縮れた糸屑上にみえる。これを嗜銀性スレッドという。進行性核上性麻痺、大脳皮質基底核変性症で比較的多く観察されるが、両疾患における嗜銀性スレッドは形態学的に異なるものである。
ニューロピル
HE染色体標本で神経細胞、アストログリア、オリゴデンドログリアの間にある均一な場所をニューロピル(neuropil)という。神経線維網と訳される。電子顕微鏡では神経細胞の樹状突起、それと接触する無数のシナプス、アストログリアの突起、ミクログリアの突起、さらに通過するだけの神経線維がニューロピルを形成する。ニューロピルの変化としては粗鬆化、海綿状態、ニューロピル・スレッドが知られている。
粗鬆化
粗鬆化は主観的な術後である。滑らかで均質なニューロピルが粗いガーゼのような形態を示すときに用いられる。
海綿状態
海綿状態(status spongiosus)とは中枢神経系の様々な場所に重篤な急性崩壊で起こるもので、神経細胞、グリアの障害によって生じる間隙と小孔である。光学顕微鏡でしばしば遭遇する皮質の海綿状態は大脳皮質の表層部(第Ⅱ層を中心として第Ⅰ層から第Ⅲ層の最上部)のニューロピルにできる細かい中空な孔の集合である。神経細胞の脱落の跡や神経細胞周囲腔(解剖学的には存在しない)あるいは毛細血管周囲なども海綿状態という言葉の中に含まれている場合もある。原因として様々な原因による脳浮腫、代謝性脳症、虚血性障害、神経変性疾患などがあげられる。特にクロイツフェルト・ヤコブ病、レビー小体型認知症の内嗅領皮質、ピック病、認知症を伴うALS、大脳皮質基底核変性症などで認められる。
ニューロピル・スレッド
ニューロピル・スレッド(neuropil threads)はニューロピルの変化ではなく、神経細胞や樹状突起と関係した変化である。特にアルツハイマー病の灰白質で広く分布する糸屑状の構造である。その密度は認知症の程度や神経原線維変化の数に相関する。ニューロピル・スレッドは神経細胞の樹状突起に由来する。アルツハイマー病に特異的ではなく、大脳皮質基底核変性症、進行性核上性麻痺、ピック病などでも認められる。
髄鞘
髄鞘とは軸索のまわりを囲っている被覆部である。オリゴデンドログリアは1つの細胞から多数の髄鞘節が形成される。一方、末梢神経のシュワン細胞では1つの細胞から1つの髄鞘節が形成される。また脳神経は末梢神経であるが、視神経の髄鞘はオリゴデンドログリアによって形成される。そのため多発性硬化症など中枢神経の脱髄性疾患では視神経異常を伴う。
髄鞘の病理的変化
- 髄鞘脱落
髄鞘の脱落を総称して髄鞘脱落という。変性疾患の軸索障害によって二次的な髄鞘の脱落も脱髄疾患による一次的な髄鞘の脱落も髄鞘脱落と表現される。髄鞘は脂質成分が多く、脱落すると脂肪顆粒細胞(貪食するマクロファージ)が浸潤する。髄鞘の破壊産物が塊状に凝集したものをミエリンオボイドという。
- 脱髄
髄鞘が一次的に崩壊する現象が狭義の脱髄である。軸索は比較的保たれる。斑状に脱髄が認められる時は脱髄斑という。
- 髄鞘融解
急激な電解質の変動、例えば低ナトリウム血症を急激に補正した場合は髄鞘染色の染色性の低下が認められ髄鞘融解という。
- 髄鞘形成不全
髄鞘が脱落するのではなく、髄鞘が十分形成されない場合、髄鞘形成不全という。白質ジストロフィーなどで認められる。
- 髄鞘低形成
先天的な髄鞘形成不全であり限局性皮質異形成や結節性硬化症では大脳皮質と白質の境界が不明瞭である。
- 髄鞘淡明化
大脳深部白質や半卵円中心では髄鞘染色で染色性の低下が認められることがあり髄鞘淡明化やミエリンパーラーという。グリオーシスやマクロファージ浸潤などの反応性変化を欠いている場合が多い。病理的な意義は不明である。
上衣細胞
上衣細胞は脳室壁を裏打ちする正方形、円柱状の細胞である。脳室側には線毛や微線毛がある。
ミクログリア
ミクログリアは小型で多極性の突起をもつ小グリア細胞である。胎生期の単球由来または神経外胚葉由来と考えられている。脳に何らかの障害が起こると突起が太くなり活性型になる。マクロファージに移行する。
ミクログリアの病理的変化
- マクロファージ
- 桿状ミクログリア細胞
- 神経貪食現象
- 異物巨細胞
中枢神経病理の代表的染色法
- HE染色(ヘマトキシリンエオジン染色)


病理学の基本的染色法であり、細胞質はエオジン好性に赤く染まり、核や核小体はヘマトキシリン好性に紫色に染まる。
- ニッスル染色
神経細胞に対する染色法であり粗面小胞体であるニッスル小体を色素のクレシル紫が紫色に染める。
- LFB染色(ルクソールファストブルー染色)
髄鞘染色である。脱髄疾患の分布の評価などを行うことができる。
- クリューバーバレラ染色
ニッスル染色とLFB染色を併用した染色で神経病理学で最も一般的な染色法の一つである。
- ボジアン染色
_presenile_onset.jpg)
嗜銀染色であり神経突起の評価や線維成分の評価に優れている。
- ホルツァー染色
クリスタル紫によってアストロサイトの線維成分を紫色に染色する。
- ガリアスプラーク染色(GB染色)
異常タウ蛋白の一部を染色する方法である。
ユビキチン、リン酸化タウ、αシヌクレイン、ボリグルタミン、GFAP(アストロサイトのマーカー)などの免疫染色を併用することが多い。
脳腫瘍の神経病理学
脳腫瘍は原発性脳腫瘍と続発性脳腫瘍(転移)に分けられる。脳腫瘍は発症年齢によって大きく発生率が異なり、成人では約70%がテント上に発生し転移、神経膠腫、髄膜腫、神経鞘腫が多い。小児では70%以上がテント下で起こり、外胚葉起源、毛様細胞性星細胞腫、髄芽腫、上衣腫が多い。脳腫瘍の症状は全身症状と局所症状に分かれる。全身症状とは腫瘍の増大とその周囲の浮腫によって頭蓋内圧が亢進したり、脳脊髄液の循環が直接障害されることによって生じる。具体的には頭痛、認知機能障害、人格障害、歩行障害といった症状である。局所症状としては片麻痺、失語、視野障害などが考えられる。原発性であれ転移性であれ、通常Gd増強効果が見られる。悪性度の低い神経膠腫ではGd増強効果が認められずFLAIR画像が診断の役にたつ。静脈血栓塞栓症が神経膠腫や転移性脳腫瘍では起こりやすいため注意が必要である。
星状細胞腫
WHOは星状細胞腫を組織学的な特徴に基づき4段階に悪性度を分類している。グレードIに毛様細胞性星状細胞腫、上衣下巨細胞性星状細胞腫、グレードIIにびまん性星状細胞腫、グレードIIIに退形成性星状細胞腫、グレードIVに膠芽腫が分類されている。Ste.Anne/Mayoの基準では、核の多形性、核分裂像、微小血管増生、壊死の4つの所見の有無によってgradeを決定している。またKi-67/MIB-1陽性率などは細胞増殖の指標として用いられる。
- 毛様細胞性星状細胞腫


若年者、特に幼児の小脳に発生することが多い良性腫瘍である。毛様の細長い突起を伸ばす腫瘍細胞が充実性の領域と浮腫を伴う海綿状の領域をつくる。充実性の領域にはエオジン好性のRosenthal fiberが認められる。
- 脳室上衣下巨細胞性星状細胞腫
結節性硬化症に随伴する脳室内腫瘍である。肥胖性星細胞に似た大型腫瘍細胞がと小型紡錘形細胞も介在し血管に富んだ構造となる。
- びまん性星細胞腫
30歳から50歳の男性に多く発生する。光顕所見では原線維性星細胞腫と原形質性細胞腫に分けられる。原線維性星細胞腫では細胞は楕円形核と好酸性細胞質をもち、多極性の突起を伸ばす腫瘍細胞がびまん性に増殖する。原形質性細胞腫は異型の弱い類円形核と狭い細胞質をもち脆弱な短突起を伸ばす腫瘍細胞からなる腫瘍である。
- 退形成性星細胞腫

50歳〜60歳の男性に多い。グレードIIの星細胞腫とくらべて細胞密度の増加、核異型、核細胞分裂像が顕著になっていることが特徴となる。
- 膠芽腫
.jpg)
原発性脳腫瘍の11%をしめ、60〜64歳にピークがある。より低悪性度の星細胞腫から多段階発癌の結果起こる二次性膠芽腫と原発性膠芽腫がある。ほとんどが原発性膠芽腫である。細胞密度は高く、細胞は小型類円形から多角形、さらに多核巨細胞などの形態を示す。核クロマチンは豊富で核分裂がいたるところでみられる。腫瘍細胞の配列は血管周囲性偽ロゼット、壊死巣を囲む偽柵状配列、血管内皮細胞の反応性増殖や腎糸球体係蹄様構造の形成が認められる。
- 大脳膠腫症
少なくとも3つの脳葉にわたる連続性の広範な浸潤を示し、テント下や時に脊髄にまでおよぶグリア系腫瘍である。長楕円形の核と狭い細胞質を持つ紡錐形細胞が既存の繊維構造に沿って浸潤性に増殖する。
乏突起膠腫

乏突起膠腫は神経膠腫のうち約15〜20%を占める。分化度の高い乏突起膠腫(平均生存期間は10年以上)と退形成性乏突起膠腫にわかれる。最も特徴的な組織像はほぼ一定の大きさの丸い核をもち、細胞質が明るくぬけ(perinuclear halo)細胞膜が明瞭な細胞がタイルを敷き詰めたように配列する。この配列をhoneycomb appearanceという。
原発性中枢神経系リンパ腫
腫瘍中心部では細胞密度が高く充実性増殖をしめし特徴的な組織構築を示さない。
髄芽腫
髄芽腫は小児の小脳で最も多い悪性腫瘍である。クロマチンに富む小型類円形の核と乏しい胞体から成る細胞がびまん性高密度に増殖する。
髄膜腫
髄膜腫は原発性腫瘍では最もおおk全体の32%を占める。髄膜腫は悪性度に沿って良性髄膜腫(グレードI)、異型性髄膜腫(グレードII)、悪性髄膜腫(グレードIII)に分類される。画像では髄膜腫の一部は石灰化し、強い造影効果を示す硬膜から発生した脳実質外腫瘍である。
神経鞘腫
神経鞘腫は原発性脳腫瘍の9%を占める。聴神経鞘腫が多い。細長い核をもつ紡錐形の細胞が柵状に配列する。
下垂体腺腫
下垂体腺腫は原発性脳腫瘍の9%を占める。
頭蓋咽頭腫
頭蓋咽頭腫は遺残組織から生じ典型的にはトルコ鞍上部にみられ、部分的に石灰化している。充実性腫瘍または充実性腫瘍と嚢胞性腫瘍が混在した良性腫瘍である。
転移性脳腫瘍
脳転移の原発巣で最も多いのは肺癌と乳がんである。
神経変性疾患の神経病理学
変性疾患の発生機序の一つの仮説に異常な立体構造をとった、あるいは異常に凝集した蛋白が細胞に毒性に作用し、ライソゾームやユビキチン・プレテアソーム系によって排除されないというものがある。この仮説に基づき、特異的封入体によって病気を分類することがある。
アミロイドβ蛋白

アミロイドβ蛋白が蓄積するのはアルツハイマー病である。アルツハイマー病における基本的病理変化はAlois Alzhaimer自身により1911年の論文において詳しく記載されている。アルツハイマー病脳病変の特徴として神経細胞の変性消失とそれに伴う大脳萎縮、老人斑の多発、神経原線維変化(英: neurofibrillary tangle、NFT)の多発の3つがあげられる。1980年代に老人斑がアミロイドβ蛋白(Aβ)の凝集蓄積であること、NFTが微小管結合タンパクのひとつであるタウが凝集線維化したものであることが明らかになった。FADの原因となるアミロイド前駆体蛋白遺伝子変異、プレセニリン遺伝子変異のいずれもAβの産生亢進を誘導することが判明している。アルツハイマー型認知症の生化学も参照。
- 老人斑
老人斑(senile plaque、SP)は鍍銀染色で濃染する径数10から100マイクロメートルほどの斑状の構造物である。細胞外に存在する。アルツハイマー病では大脳皮質に大量の老人斑が出現する。老人斑(senile plaque、SP)はAβの凝集蓄積により形成されるが、ある程度以上の大きさと密度をもつ蓄積の場合、老人斑内とその周囲にある神経突起の変性が生じる。変形神経突起の半数以上においてNFTとそう痒のタウの凝集蓄積が生じている。Aβの蓄積は斑状構造、綿屑のようなもの、まばらな蓄積など様々な形態をとる。Aβは分子量約4kDの小さな蛋白質で38 - 43個のアミノ酸からなり、695 - 770個のアミノ酸からなる1回膜貫通型のアミロイド前駆蛋白 (APP) からプロテアーゼにより切りだされて産出される。複数の分子種が知られており、それらの分子種は性質の違いからC末端が短いAβ40と長いAβ42に大別される。Aβの産出自体は生理的な現象である。Aβ40は水に溶けやすく、凝集、蓄積を生じにくいのに対してAβ42は凝集傾向が強い。老人斑形成の過程においてはAβ42の沈着が先行し、後の段階でそれに巻き込まれるようにAβ40も沈着すると考えられる。老人斑ではAβの蓄積と変性神経突起形成に加えてグリア細胞の反応が生じている。老人斑の中心部には活性化ミクログリアが集簇をなし、周辺部には活性化アストロサイトが老人斑を取り囲むように存在する。老人斑は慢性炎症性変化と考えられている。アストロサイトはミクログリアと同様にAβの除去や炎症性因子の産出、それに老人斑という慢性炎症性病変を周囲の脳組織から隔離する機能を果たしていると推測される。
アミロイド沈着は基本的にアミロイド密度が高い核(または芯)をもつ典型的老人斑 [注釈 1]と密度の低いびまん性老人斑[注釈 2]の2つのタイプがあることが知られている。
老人斑はAβ沈着からなる好酸性斑状構造物で、細胞外マトリックス、すわなちニューロピルに形成される。典型的老人斑(ないし古典的老人斑)は中心部のコアとその周囲の腫大した軸索(dystrophic neurite、DN)を有する。原始老人斑はAβ沈着を有し、コアは明らかでないものの、DNを伴っている。典型的老人斑と原始老人斑はDNを有する特徴よりneuritic plaque(NP)と呼ばれる。NPは特に生理的な老化との比較においてアルツハイマー病病理を大きく特徴づけるものである。DNはp-tau染色で陽性である。びまん性老人斑はAβ沈着を有するが、その輪郭は不明瞭でコアはみられず、またDNやその他のp-tau病変を伴わないもので生理的老化でもひろく認められる。
- 神経原線維変化 (NFT)
神経原線維変化(NFT[注釈 3])は神経細胞の細胞体に生じる繊維状の凝集体で、その微細構造は長径10ナノメートルのフィラメントが2本ずつらせん状のペアを作った線維の集合体であり、規則的なくびれ構造をもつ。らせん状のペアを作った線維を対らせん線維(PHF[注釈 4])という。PHFはタウが重合してβシート構造を形成することによって生じる。PHFを構成するタウは通常のタウと異なり過剰にリン酸化を受けている。PHFは細胞質内にありながらプロテアーゼ耐性である。主要構成蛋白であるタウは神経細胞軸索における微小管の安定化に関わる因子である。タウのC末端には微小管の結合部位がありリピート配列がある。ADでは3Rと4Rの両方のアイソフォームからなる。NFTは鍍銀染色陽性でありガリアスブラーク染色が用いられることが多い。タウの凝集や蓄積の過程で分子構造が変化するため免疫染色ではエピトープが消失することもあるので注意が必要である。
NFTはまずは神経細胞の胞体内に形成されintracellular NFT (iNFT)とよばれる。海馬や海馬傍回のiNFTは火炎状であり、その主成分はp-tauである。神経細胞の死滅によりNFTが細胞外に沈着すると、好酸性が増してextracellular NFT (eNFT)、またはghost tangleになる。また背景には変性した樹状突起(HE染色では確認できないニューロピルスレッド)が多数見られp-tau陽性である。また海馬では平野小体や顆粒空胞変性も認められる。これらのアルツハイマー病病変は灰白質を主体とするもので、末期においても白質はよく保たれている。進行性核上性麻痺、大脳皮質基底核変性症、多系統萎縮症では白質にも第一義的病変が認められることと対照的である。
- アミロイドアンギオパチー
アルツハイマー病では脳実質に出現する老人斑に加えて、脳血管壁にコンゴーレッド陽性になるアミロイドが沈着する脳血管アミロイドーシス(脳アミロイドアンギオパチー、CAA)を併発する率が高い。これが脳出血を起こす原因となっている。特に家族性アルツハイマー病でAPP遺伝子内にAβの内部配列に変異が起こると脳アミロイドアンギオパチーを多発するケースが多い。血管アミロイドは中膜と外膜の間の基底膜にまばらに沈着し、進行的に全周性に沈着がみられ平滑筋細胞の消失をもたらす。
- アルツハイマー型認知症病理の時系列
病理の時系列解析は主にダウン症患者由来の剖検脳を用いた検討で明らかになった。ダウン症患者ではアルツハイマー病の病理変化が30代ころから出現し年齢を重ねると共に進行する。アミロイドの沈着は30代から始まり、40歳以降で神経原線維変化が出現する。また神経原線維変化がある場合は必ず老人斑を伴うことが明らかになった。ダウン症は第21染色体トリソミーであるため第21染色体に存在するAPP遺伝子が通常の1.5倍存在する。その結果APPのmRNAも通常人よりおおく早発認知症になると考えられている。
- 合併病変
ADに本質的な病変は神経細胞消失、老人斑、NFTである。しかし偶然の合併で考えられる以上にレビー小体、TDP-43など他の疾患に特徴的な病変を合併し、臨床像に影響を与える。病理機序の一部は共通していると考えられる。なお病理診断でレビー小体型認知症とADが合併した場合はADらしさの高低を優先して判断することになっている。アルツハイマー病とレビー小体型認知症の合併例は非常に多い[1]。臨床診断がアルツハイマー病であった症例の32~40%で病理診断はアルツハイマー病とレビー小体型認知症の合併であったという報告がある。また臨床診断でレビー小体型認知症であった症例の32~52%で病理診断はアルツハイマー病とレビー小体型認知症の合併であった。また病理診断でアルツハイマー病とレビー小体型認知症の合併と診断された例の61%は臨床診断がアルツハイマー病であった。
- 病理診断
アルツハイマー病で認められる病理変化は生理的な老化でも出現するため定性的診断は困難である。そのため半定性・半定量的な病理診断が行われる。
- Braak NFT stage
Braakによるstage分類は神経原線維変化(NFT)を用いたBraak NFT stageと老人斑(SP)を用いたBraak SP stageがある。鍍銀染色をもとにしたもの[2]と、p-tau(AT8)免疫染色をもとにしたもの[3]とがあるが、一般的にはp-tau免疫染色を使うことが実際的と思われる。病変は側頭葉内側より始まり(stageⅠ~Ⅱ)、海馬・辺縁系に及び(stageⅢ~Ⅳ)、最後に新皮質に進展する(stageⅤ~Ⅵ)。Primary cortex(一次運動野、体性感覚野、視覚野、聴覚野)が一番最後に障害される(stageⅥ)。
- Braak SP stage
Braakによるstage分類は神経原線維変化(NFT)を用いたBraak NFT stageと老人斑(SP)を用いたBraak SP stageがある。Braakらは鍍銀染色を使用している[2]が一般的にはAβ免疫染色を使うことが実際的と思われる。老人斑の沈着は新皮質下面(前頭・側頭・後頭葉)から始まり(stage A)、次に新皮質(連合野)全体に及び、かつ海馬が障害されはじめ(stage B)、最後にprimary cortex(一次運動野、体性感覚野)が障害される(stage C)。大脳基底核や視床への沈着がみられるのもstage Cである。p-tau病変とは異なり、全stageを通して海馬での老人斑沈着は比較的軽度にとどまる。
- CERAD score
Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) score[4]は生理的老化にはみられにくく、AD組織の大きな特徴であるneuritic plaqueを半定量化し(NP score)、死亡時年齢による重みづけを行い、組織学的なアルツハイマー病らしさを判定する。CERAD scoreを作成したMirraらはneuritic plaque(NP)可視化のためにBielschowsky鍍銀染色ないしはthioflavine S蛍光染色を使用しているが、一般的にはAβ免疫染色染色(典型的老人斑を数える)もしくはp-tau免疫染色(p-tau陽性NPを数える)を使うことが実際的と思われる。
- Thal SP(Aβ) phase
Thal SP(Aβ) phaseまたはタールらの基準[5]はBraak SP stageとは異なり、大脳のみならず脳幹や小脳を含めて、その老人斑沈着を鍍銀染色ないしAβ免疫染色により分類するものであるが、Aβ免疫染色を使うことが実際的と思われる。5つのphaseに分けられる。Phase1では老人斑は新皮質にとどまる。Phase2では老人斑は新皮質だけではなく不等皮質(海馬や帯状回など)に分布する。Phase3ではさらに間脳(視床や視床下部)や線条体に分布する。Phase4ではさらに脳幹諸核(黒質、赤核、下オリーブ核など)に分布する。Phase5ではさらに小脳・脳幹諸核(橋核、青斑核など)に分布する。
- ABCスコアによるアルツハイマー病病理診断基準
ABCスコア[6]はアルツハイマー病の進展様式を総合的に組み込んだもので組織学的なアルツハイマー病らしさを4つのカテゴリー(not、low、intermediate、high)に分類する。そしてintermediateまたはhighが、その認知症を説明するに十分なアルツハイマー病理と考えられる。NIA-AAの病理診断の基準もABCスコアである。合併病変も合わせて評価する。
タウ蛋白
進行性核上性麻痺、皮質基底核変性症などがタウオパチーとして知られている。
タウ蛋白
1975年タウは神経系に特異的に発現する微小管結合蛋白質として発見された。微小管はα、βチューブリンのヘテロ二量体からなる主要な細胞骨格のひとつと考えられている。タウは細胞内において微小管の重合促進および安定化、細胞骨格構造の形成、維持に重要な役割を果たし、その機能はリン酸化(大きな電荷によるコンホメーション変化)によって調節されている。タウ遺伝子はヒトでは17番染色体上17q21.2に存在し、16個のエクソンからなる。タウは単一遺伝子から転写されたpre-mRNAが選択的スプライシングされることで6つのアイソフォームが発現する。エクソン2,3,10の選択的スプライシングの結果アミノ酸352〜441個の6つアイソフォーム、即ち352(0N3R)、381(1N3R)、383(0N3R)、410(2N3R)、412(1N4R)、441(2N4R)ができる。Rはタウのカルボキシル基末端側の微小管結合部のリピート数を示す。微小管結合部はエクソン9〜12にコードされておりエクソン10を含む4Rタウとエクソン10を含まない3Rタウに分けられる。タウの微小管結合能は4Rの方が大きくN末端の配列は影響しない。NはタウのN末端部位に存在するプロジェクション領域と言われる部分のプロフィールであり微小管の間の間隔を決定している。エクソン2.3の有無によって決定されエクソン2,3ともに認められないと0Nであり、エクソン2がある場合は1N、エクソン2.3ともにあれば2Nと分類される。ヒト胎生期〜新生児期は352(0N3R)のみ発現するが成人では6つのアイソフォームすべてが発現する。これは微小管ネットワークのダイナミクスを保つ上で3Rタウによる微小管形成が必要であり、安定な微小管ネットワークを保持するには4Rタウによる微小管形成が必要である可能性が示唆されている。神経細胞内線維状封入体を形成するタウのアイソフォームは各疾患によって異なり主に3R型、主に4R型、あるいは3R、4R両者が同じ比率で含まれるタイプに分類される。3R型にはピック病、4R型には進行性核上性麻痺、皮質基底核変性症など、両者にはアルツハイマー病などがある。
進行性核上性麻痺
進行性核上性麻痺(progressive supranuclea palsy PSP)は1964年にsteele JC、Richardson JC、Olszewski Jの3人によって報告された疾患である。原著では7人の剖検例を含む9例のPSP患者の報告がされている。その臨床的特徴としては垂直性注視麻痺、偽性球麻痺、項部ジストニア、認知症、姿勢保持反射障害があげられている。10万人あたり6人程度である。臨床診断基準を満たすものでもいくつかの亜型があることが知られている。
- Richardson症候群
早期からの易転倒性、垂直性核上性眼球運動障害を示す典型的PSP。
- PSP-P(PSP-parkinsonism)
発症時に左右差、振戦がみられL-DOPAに当初反応しパーキンソン病と紛らわしい群。
- PAGF(pure akinesia with gait freezing)
歩行や書字、発語のときのすくみを主症状として、筋強剛や振戦がみとめられずL-DOPAに対する反応性がないもの。すくみが他の神経症候より長時間先行し罹患期間は平均13年と長い。
- CBS(corticobasal syndrome)
PSPの中には大脳皮質基底核変性症(CBD)様の症状、即ち左右差のある錐体外路症状(パーキンソニズムやジストニア)、皮質症状(失行、失語、皮質性感覚障害)を示す報告例も認められている。
- 肉眼所見
中脳と橋被蓋の萎縮、中脳水道及び第三脳室の拡大。黒質の褪色と萎縮が高度であるが青班核の褪色は軽度。
- 組織所見
globose型アルツハイマー神経線維変化(NFT)の出現を伴う神経細胞の脱落とグリオーシスを基底核、視床、脳幹部に認め、淡蒼球、視床下核、黒質に最も強い。GB染色(ガリアスプラーク染色)や免疫染色ではNFTは神経細胞脱落の強い領域を超えて大脳から脊髄まで広い範囲で観察される。タウの蓄積したグリア細胞の封入体(glial fibrillary tangles:GFT)が出現するのはCBDとの共通点である。GFTにはオリゴデンドログリア胞体内のcoiled body、アストロサイトに形成されるtuft-shaped astrocyte、有髄線維内のargyrophilic thredから成りtuft-shaped astrocyteはPSPの病理診断指標の一つである。tuft-shaped astrocyteは側枝を有さない細い突起が中心部から放射状に配列する形態を示すものであり、PSPでは中心前回を主体に前頭葉、頭頂葉に分布する他、基底核や脳幹に認められる。原則としてはPSPはtuft-shaped astrocyteを、CBDはastrocytic plaqueを示し病理学的には通常は両者の合併は見られない。しかし両者の共存例の少数報告例は存在する。
皮質基底核変性症
皮質基底核変性症(CBD)は神経細胞とグリア細胞の双方におよぶタウオパチーであり、astrocytic plaqueは診断的価値を有する構造物である。CBDでは大脳皮質と皮質下神経核(特に黒質)に神経脱落が認められる。大脳皮質にballoned neuronが出現していることに意味があり、その数は問わない。大脳上面に出現していることが重要であり、balloned neuronが辺縁系に限局している場合はAGD(嗜銀顆粒性認知症)の合併が疑われるとされている。PSPの鑑別法としては先にあげたPSPはtuft-shaped astrocyteを、CBDはastrocytic plaqueの他、PSPでは淡蒼球、視床下核、黒質の病変が必発でありこれに小脳歯状核、脳幹被蓋の加わるが、CBDでは淡蒼球と黒質の病変が高度であるが線条体は中等度、視床下核と小脳歯状核の変化は軽度とされている。
- 肉眼的所見
大脳皮質の限局性萎縮、淡蒼球の萎縮と黒質の色素脱失。
- 組織所見
Nissl顆粒の融解と胞体の腫大を示す神経細胞(balloned neuron)が認められる。萎縮を呈する大脳皮質の3層および5,6層に認められる。GB染色や免疫染色では神経細胞、グリア細胞ともにタウの蓄積が認められる。診断上有用なものとしてはastrocytic plaqueがあげられる。これはアストロサイトの遠位部にタウが蓄積したものであり短い突起状の構造物が集合して1つの班を形成する。老人斑と異なりアミロイドの沈着は認められない。その他、オリゴデンドログリア胞体内のcoiled body、有髄線維内のargyrophilic thredが認められる。
トピックス
- アルツハイマー病に関して
1980年代後半に異常リン酸化されたタウがアルツハイマー病の脳において神経細胞内に蓄積する神経原性変化の構成単位であるpaired helical filaments(PHF)およびstraight filamentsの主要構成成分として同定された。現在はpaired helical filaments、straight filamentsをまとめてタウ線維という。その後多くの神経変性疾患において主要な病理蓄積物の構成成分であることがわかりタウオパチーという概念が出来上がった。
- FTDP-17
1998年に前頭側頭型認知症のひとつであるFTDP-17においてタウ遺伝子内に変異が見つかった。タウの機能異常が神経細胞死および認知症を直接誘導する可能性が示唆された。
- PSP、CBDの危険因子
タウ遺伝子において連鎖不均衡部位があり、この部位のSNPおよび転座によってH1、H2の2つのハプロタイプに分けられている。H1ハプロタイプはPSP、CBDの危険因子と考えられている。
αシヌクレイン

ポリグルタミン
TDP-43
基底核の病理学
多系統萎縮症では被殻の褐色萎縮、DRPLAでは淡蒼球外節の萎縮、MJD(SCA3)では淡蒼球内節の萎縮が認められる。ハンチントン病では尾状核の萎縮が、進行性核上性麻痺では淡蒼球とルイ体(視床下核)の萎縮が大脳皮質基底核変性症では淡蒼球と視床の萎縮が認められる。
小脳の病理学
小脳皮質の病理
加齢性変化
小脳は脳幹とともに重量の減少が大脳に比べて小さい、20歳代の平均脳重量に対して100歳代では大脳は20~25%減少しているのに対して小脳や脳幹は10~15%程度の減少となる。肉眼的には加齢性変化は小脳虫部で顕著である。特に小脳第一裂より上面の前葉である中心小葉や山頂で目立つ。
マクロ病変
キアリ奇形といった奇形や脳ヘルニアや交叉性小脳萎縮(crossed cerebellar atrophy)がよく知られたマクロ病変である。遠隔機能障害(diaschisis)の一種として示されることもある。交叉性小脳萎縮症とは広範な一側性の大脳病変から長い年月を経て反対側の小脳が萎縮する現象である。Urichらは病理発生機序の立場から交叉性小脳萎縮を3つのタイプに分けている。それは
- 前頭・側頭橋路の病巣に続いて起こる橋核の順行性経ニューロン変性と中小脳脚の萎縮による小脳皮質に病変を伴わない小脳半球の萎縮
- 反対側の橋核の順行性経ニューロン変性による顆粒細胞層萎縮が顕著な小脳萎縮
- てんかん発作によると考えられる小脳萎縮
びまん性のミクロ病変
大脳皮質と同様に小脳皮質も層に強調された変化を示す。プルキンエ細胞の変化が中心となることが多く、変性疾患では小脳背側部で虫部と傍虫部半球に病変が強調される傾向がある。
- 分子層
分子層はHE染色ではエオジンに染まる細かい網目状のニューロピルと小型の籠細胞(basket cells)、星状細胞(stellate cells)からなる。プルキンエ細胞の樹状突起や顆粒細胞の軸索が鍍銀染色で確認できる。分子層では固有の疾患は知られていない。前述のプルキンエ細胞の樹状突起や顆粒細胞の軸索があるためプルキンエ細胞層や顆粒細胞層に変化が生じると分子層でアストログリアが造成することが多い。プルキンエ細胞の樹状突起が限局性に膨らみ突起が出ているように見えるカクタスが認められることもある。カクタスは代謝性疾患や発達障害で有名だが多系統萎縮症、皮質性小脳萎縮症、CJDなどで認められ疾患特異性はない。
- プルキンエ細胞層
分子層の下端に大きなフラスコ型の細胞が1列並んだプルキンエ細胞層がある。その樹状突起は分子層の中で扇のように平面的に枝分かれする。その面は小脳回に対してほぼ直角である。プルキンエ細胞は虚血に対して非常に脆弱な細胞であるため、死後変化や死戦期の浮腫かどうかを区別するためにベルグマングリア(小脳のアストログリア)の増殖を確認する。死後変化や死戦期の浮腫ではプルキンエ細胞層が海綿状に離開し、プルキンエ細胞は消失しているが、アストログリアの反応はみられない。
プルキンエ細胞の胞体は分子層にある籠細胞の突起によって取り囲まれている。正常ではその他の神経線維も同時に染まるためバスケットの部分はわかりにくいがプルキンエ細胞が脱落するとempty basketsという所見で確認ができる。トルペドはプルキンエ細胞の最も近位部の軸索に生じたスフェロイドであり顆粒細胞層内で認められる。プルキンエ細胞の障害を示唆する所見だが疾患特異性はない。多系統萎縮症では多数認められることがあるが遺伝性脊髄小脳変性症では遭遇することは稀である。
皮質性小脳萎縮症は病理学的には下オリーブ核-小脳虫部という登上線維系に限局する病変を示す。多系統萎縮症やマチャド・ジョセフ病では主たる病変が小脳へ入力する苔状線維系の変性であること、病変がそれ以外にも複数の部位で認められる点が皮質性小脳萎縮症とは異なる。またアルコール性小脳萎縮症や自己免疫性小脳失調症や傍腫瘍性神経症候群では病巣が不連続的、あるいは解剖学的な部位と無関係な病変の強弱が認められる。
- 顆粒細胞層
皮質の中で最も厚く見える層が顆粒細胞層である。円形でクロマチンに富む小型の細胞核が密集しているため、HE染色標本の弱拡大像では顆粒層全体が青紫色にみえる。顆粒細胞が脱落する場合は白質側から消失することが多い。
限局性のミクロ病変
小脳の限局性病変では梗塞が多い。
小脳髄質の病理学
変性疾患、白質ジストロフィー、脱髄性疾患、腫瘍で小脳髄質(白質)に病変が認められる。
変性疾患
多系統萎縮症は小脳の割面は白質の萎縮が強いため皮質が相対的に大きく見える。これは橋核(苔状線維系)のみならず下オリーブ核(登上線維系)から小脳に入る神経線維の変性がプルキンエ細胞から歯状核に向かう神経線維の変性を凌駕しているからと考えられている。その小脳白質の割面は非常に特徴的であり肉眼的には境界不鮮明な斑状の白い部分とやや褐色を帯びた部分が混在している。多系統萎縮症では変性が歯状核を超えることはない。一方でマチャド・ジョセフ病では歯状核門から上小脳脚に線維性グリオーシスが広がる。
白質ジストロフィー
副腎白質ジストロフィーでは病変が小脳白質から橋底部、中小脳脚、下オリーブ核、下小脳脚などに左右対称性にひろがる。アレキサンダー病ではローゼンタール線維が出現する。
脱髄性疾患
多発性硬化症などで脱髄性病変を伴うことがある。
腫瘍
小児腫瘍が小脳髄質に起こりやすい。髄芽腫は小脳虫部下面に好発する。
小脳核の病理学
小脳核は第四脳室の天井付近に室頂核、球状核、栓状核、歯状核の4つの神経核があり、発生学的には下オリーブ核と同じ起源である。プルキンエ細胞はこの小脳核のいずれかに投射線維を送る。歯状核が最も大きく、歯状核以外に選択的に障害を示す疾患が知られていないため神経病理学では歯状核に注目する。歯状核の入力線維は外側から入る。歯状核の内側部は上小脳脚に向かって歯状核門が開いており、歯状核の出力線維は歯状核門と上小脳脚を通る。
血管障害
歯状核は循環障害の影響を受けやすい。低酸素脳症では歯状核の脱落が認められる。小脳出血の好発部位でもある。赤核と同側の下オリーブ核、それに反対側の小脳を結んだ線をギラン・モラレの三角という。この三角の一部が梗塞や外傷で切断されると下オリーブ核に肥大が生じることがある。とくに病巣に歯状核が含まれている時に観察されることが多い。
変性
歯状核の変性には神経細胞の膨化、グルモース変性、神経細胞の変性、脱落が知られている。ペラグラ脳症やクロイツフェルト・ヤコブ病では歯状核のリポフスチン顆粒の沈着がほとんどないにも関わらず神経細胞がふくらんでいることがある。日本ではグルモース変性といえば小脳歯状核にみられる変化を示す場合が多い。HE染色では好酸性を呈する雲状の構造物が集積する。プルキンエ細胞軸索終末の前シナプス変化と考えられており、小脳歯状核細胞周囲で軸索末端部で無髄線維が増加(発芽)と考えられている。プルキンエ細胞が高度に脱落している場合はグルモース変性が認められない。小脳遠心系変性を示す所見であり進行性核上性麻痺や歯状核赤核淡蒼球ルイ体萎縮症などで特徴的に認められる。歯状核の神経細胞の変性・脱落では歯状核自体に生じる一次性脱落と小脳皮質の病変の二次性脱落に分けることができる。一次性脱落は進行性核上性麻痺と歯状核赤核淡蒼球ルイ体萎縮症、フリードライヒ失調症、ミトコンドリア病のMERRF、ラフォラ小体病などでみられる。進行性核上性麻痺と歯状核赤核淡蒼球ルイ体萎縮症ではグルモース変性が非常に特徴的であるが、高度な神経細胞脱落もおこる。ミエリンの淡明化が主に歯状核門に生じ、線維性グリオーシスで置換されている。さらに歯状核門から上小脳脚にもグリオーシスが認められる。マチャド・ジョセフ病では神経細胞の脱落に比べてミエリンの淡明化が歯状核の外側部と内側部の双方に認められる。フリードライヒ失調症は歯状核の神経細胞脱落と上小脳脚の変性、淡蒼球やルイ体変性を伴うことがある。またミトコンドリア病のMERRFでは小脳歯状核、下オリーブ核、黒質、基底核などに著しい萎縮、神経細胞の脱落やグリオーシスがみられる。また大脳基底核や大脳白質の血管に石灰化が生じる。ラフォラ小体病の小脳ではプルキンエ細胞と顆粒細胞の中等度脱落に加えて歯状核の神経細胞が高度に脱落する。二次性脱落は自己免疫性小脳失調症、皮質性小脳萎縮症、アルコール性小脳萎縮症、多系統萎縮症などでみられる。一次性脱落に比べると神経細胞の脱落は高度ではなく萎縮が中心となる。アストログリアの増殖を伴うが線維性グリオーシスが特徴とされる。自己免疫性小脳失調症、皮質性小脳萎縮症、アルコール性小脳萎縮症などプルキンエ細胞が標的となっている疾患では、プルキンエ細胞の軸索の変性が歯状核に収斂するため歯状核外側部に接する白質にマクロファージが集簇する。特に傍腫瘍性小脳変性症ではマクロファージが歯状核外側部のみならず途中の白質にもみられることがある。多系統萎縮症でも歯状核は萎縮が主体である。歯状核門に白質病変がおよぶことは非常に稀で上小脳脚は保たれる。
小脳萎縮のまとめ
小脳萎縮には3つの表現型が知られている。特に小脳皮質変性(プルキンエ細胞型)と歯状核変性(歯状核型)は明らかに区別できる。プルキンエ細胞型の代表例は多系統萎縮症(MSA-C)であり、小脳半球の白質、プルキンエ細胞が脱落し、歯状核、歯状核門は保たれる。顆粒細胞型の代表例はメンケス病やGM2ガングリオシドーシスといった代謝性疾患である。メンケス病は銅の細胞内代謝障害である。プルキンエ細胞も脱落するが顆粒細胞の脱落が著しいのが特徴である。MELASをはじめとしたミトコンドリア病も小脳萎縮を示すことで有名である。画像上は小脳萎縮を示すが明らかな小脳性運動失調を認めないことも多い。顆粒細胞型ではプルキンエ細胞が限局的に腫大したカクタスやヒトデ小体が認められることがある。ヒトデ小体は樹状突起の遠位部の腫大であり分子層にみられるが、カクタスはプルキンエ細胞層で認められる。歯状核例の代表例はマチャド・ジョセフ病であり歯状核、歯状核門、上小脳脚が脱落し、歯状核はミクロ的にはグルモース変性像を呈する。プルキンエ細胞は保たれる。グルモース変性では好酸性、嗜銀性のもやもやした無構造の物質と顆粒状あるいはリング状の物質が歯状核の神経細胞体や樹状突起の周囲に巻き付くものでプルキンエ細胞の軸索末端変化とされている。マチャド・ジョセフ病では淡蒼球内節の萎縮も特徴的である。ミトコンドリア病のMERRFは小脳歯状核にも著しい萎縮がみられる。
Remove ads
末梢神経病理
要約
視点
末梢神経の生検では腓腹神経が選択されることが多い。生検後運動麻痺をきたさないこと、遠位部にあるためポリニューロパチーの病変が検出しやすいこと、位置が同定しやすく圧迫などの外傷から守られていること、神経伝導速度検査と対比が可能なことからである。血管炎性ニューロパチーやサルコイドーシスを疑う場合は筋生検を同時に行うため浅腓骨神経と短腓骨筋の同時生検や高位腓腹神経と短腓骨筋の同時生検がされることがある。検体はHE染色のためのホルマリン固定、エポン包埋を行うトルイジンブルー染色や解きほぐし像のためのグルタールアルデヒド固定、免疫染色のための凍結標本などの作成のために分割することが多い。標準的には凍結標本、エポン包埋標本(トルイジンブルー染色で光顕標本、また電顕標本にもなる)、ときぼぐし標本の4種類を作成する。
末梢神経病理の代表的染色法
- トルイジンブルー染色
有髄線維の評価を行うのに最も適した染色法と考えられ、末梢神経病理では最も一般的な染色法の一つである。エポン包埋で行う。この標本で軸索変性、脱髄に関して多くの情報が得られる。急性の軸索変性であるミエリン球(myelin ovoid)、慢性の軸索変性であるクラスター化、急性の脱髄をしめすNaked Axinや反復する脱髄であるオニオンバルブ(onion bulb)が評価できる。
- HE染色
パラフィン包埋切片である。個々の神経の観察には適さないが神経生検が特異的診断に直結する疾患では極めて有効である。アミロイドや血管炎、炎症細胞浸潤、サルコイド結節、ハンセン病におけるらい菌などの評価を行うことができる。
- 免疫染色
血管炎や悪性リンパ腫、抗MAG抗体ニューロパチーなどで用いられる。
- ときほぐし像
軸索変性や脱髄など個々の有髄線維の病的プロセスが検討できる。急性、亜急性の軸索変性ではミエリン球の連鎖が観察でき、脱髄と再髄鞘化過程として知られるランビィエ絞輪の染色性の低下(節性脱髄や再髄鞘化を示す所見)やランビィエ絞輪間距離のばらつきがみられる。
神経生検
体位はうつ伏せ、あるいは手術肢側を上にした斜め45°仰臥位または側臥位で手術肢を膝で90°屈曲した形のいずれかで行う。腓腹神経生検でも高位の生検法を用いれば短腓骨筋の採取も可能である。腓腹神経神経生検、高位腓腹神経生検、浅腓骨神経生検などの方法が知られている。
- 高位腓腹神経生検
静脈確保後外踝後方の外踝上縁より約2横指上方、アキレス腱との間の部位を中心に剃毛し、消毒する。アキレス腱と平行に3〜4cm程度切開をいれる。結合組織を鈍的に剥離していくと、切開創と平行に走行する直径2〜3mmの2本の管状構造物が認められる。これが小伏在静脈と腓腹神経である。両者の鑑別は比較的困難である。小伏在静脈は腓腹神経よりも表面に近いところを走行すること、神経には絹のような光沢があり、数本の縦に走行する数本の神経束の束として認められること、血管は直角に分枝するが神経は直角に枝を出すことはないといったことで区別を行う。腓腹神経を確認したら小伏在静脈を十分に剥離し、神経が直視下に入るようにする。神経をつまんだり、圧迫しないように注意し、糸をゆるくかける。その後近位端を切断する。この時に電撃痛が認められる。その後遠位端の切断を行う。神経は3cm以上採取する。止血を確認したら皮膚縫合を確認する。 短腓骨筋の生検を行いたい場合は神経生検後の止血を確認した後にさらに結合組織を鈍的に剥離し深部に進む。腓腹神経が腓骨とアキレス腱の中間部よりもアキレス腱よりに位置するのに対して、短腓骨筋はやや腓骨寄りにあることに留意する。筋膜を見つけたらメスで切開し、通常の筋生検と同じ方法で筋肉標本を切除する。創部は密閉されていれば消毒、ガーゼ交換は3日おき位で十分である。生検施行後は2〜3日は術肢免荷であり移動は車椅子である。その後より荷重可能、シャワー可能となり、10日程度で抜糸可能となる。その後入浴可能とする。術後の関節を動かした時に電撃痛が認められることがある。感覚鈍麻、異常感覚が腓腹神経領域の後遺症として残る。後遺症の分布は縮小傾向となり1年後に強い症状を訴えるものは10%以下とされている。
末梢神経の正常組織
腓腹神経の検体では正常では有髄線維、無髄線維、シュワン細胞、結合組織が認められる。有髄線維の大部分は感覚線維であり、長径分布は大径(直径7〜12μm)、小径(長径1〜4μm)の2峰性であり、密度は6000〜10000/mm2の間にある。腓腹神経では大径線維が35〜45%を占め、小径線維が55〜65%を占める。無髄線維は感覚線維と交感神経節後線維で割合は7対3である。1峰性の分布(長径0.1〜2.0μm)をとり密度はほぼ20000〜40000/mm2である。軸索変性後はsproutsが多数無髄線維として認められるため線維密度が増加する。無髄線維は有髄線維の約4倍存在する。髄鞘はG-ratioで評価される。G-ratioは神経全体の直径に対する軸索の直径であり0.6〜0.7くらいで良好な神経伝導がえられるとされている。しばしば神経周膜下に線維芽細胞や基底膜の残骸やコラーゲン繊維などを含むルノーボディが認められるが病的異議は乏しいとされている。
また腓腹神経では加齢性変化も知られている。加齢にともない結合組織が増えて神経内鞘の面積が増加し、実数は減っていないのに密度が低下する。大径有髄線維が高齢になると減少する。70歳代は30歳代の70%に低下するといった報告や80歳代は20歳代の54%程度といった報告もある。また加齢に伴い神経周膜基底膜が肥厚し、内鞘血管のヒアリン化が目立つ傾向がある。神経病理学
髄鞘の成分は中枢神経と末梢神経で異なる。中枢神経のミエリンの主成分はミエリン塩基性タンパク質(MBP)とミエリン・プロテオリピドタンパク質(PLP)であるが、末梢神経のミエリンの主成分はP0とP2である。
末梢神経の障害
末梢神経病理学の重要な点のひとつに有髄線維の変化の1次的な原因が軸索変性か髄鞘なのかを鑑別することである。軸索と髄鞘の依存関係のためこの鑑別はしばしば困難である。軸索が障害を受けると、軸索から様々な栄養因子を供給されていた髄鞘が維持できなくなって崩壊し髄球となった処理される。また脱髄では髄鞘に保護されていた軸索が露出することになり、再びシュワン細胞による髄鞘化を受けないでいると萎縮したり消失したりする。また神経生検特有の問題として生検部位の軸索変性が近位部で生じた脱髄の二次的変化による場合のこともある。
軸索障害
軸索内では蛋白の合成ができないため、細胞体からの軸索輸送がその維持に重要である。軸索の形態的変化は輸送の流れが途絶えたためにその遠位部では細胞骨格や小器官の消失が起こる。それにひき続いて軸索の破壊、髄鞘構造の破壊が起こり、髄鞘の塊(髄球)となりマクロファージによる処理がされる。軸索とともに髄鞘成分も取り除かれた後、シュワン細胞が残存し、軸索の再生に備える。軸索の伸長はgrowth coneと呼ばれる先端の膨らんだ部分が誘導する。多数の軸索の芽が分枝し、かつて1本の有髄線維を取り巻いていた基底膜内に入る。ある分枝は長軸方向からそれたり反転したりして接合先を失う。したがって初期の再生繊維は通常は3本以上の無髄線維や薄い髄鞘をもった線維が共通の基底膜に覆われている。
軸索障害の初期では髄鞘軸索変性が光学顕微鏡でオボイド(ovoid、ミエリン球)として認められる。これは壊れた髄鞘に囲まれた軸索の破片である。圧迫によるアーチファクトとしばしば鑑別は困難である。オボイドは電子顕微鏡やときほぐし像で最も評価ができる。慢性期または進行期の軸索変性では有髄軸索の減少と神経内膜の結合組織の増加である。再生クラスターの存在は、その背景にある病理変化が軸索変性による仮定の良い証拠となる。ときほぐし標本によって神経変性と再生の異なる病期を示し、病変が進行中であることを示すこともできる。軸索径の変化がしばしば診断の手がかりになる。軸索径は軸索にふくまれるニューロフィラメントと微小管の数と相関がある。軸索萎縮はほとんどニューロフィラメント生成の減少よっておこる。大径線維がニューロフィラメントに最も富むため最も病変を認めやすい(神経径依存性の脆弱性)。神経障害が長期に及ぶと二次性脱髄が起こり、重度になると原発性の慢性脱髄に過程が似ることもある。高齢者の軸索萎縮はシャルコーマリートゥース病、尿毒症性ニューロパチー、糖尿病性ニューロパチー、骨髄腫に関連したニューロパチー、種々の中毒性ニューロパチーで認められる。局所性あるいは多巣性のニューロフィラメントやその他の細胞小器官の蓄積によっておこる軸索腫大(スフィロイド)は遺伝性巨細胞性ニューロパチーやn-ヘキサンによる中毒性ニューロパチーで認められる。
軸索変性は次に示す3つのタイプが知られている。どのタイプでも変性の過程が運動軸索に影響すると最終的には筋肉の脱神経をきたす。
- ワーラー変性
ワーラー変性は神経離断に対する軸索遠位部の反応である。ヒトでは局所の虚血や圧迫などが対応する。早期には、形態学的には軸索とその髄鞘の破壊が特徴である。続いて修復期では、シュワン細胞の基底膜カラ形成されるくだの中にシュワン細胞が増殖する。管を形成するシュワン細胞の集まりはビュングナー帯を構成している。軸索の再生は切断された神経の近位断端から軸索の萌芽(sprouting)を通じて軸索の切断とほぼ同時にはじまる。進行は1日に1〜3mmと遅い。これらの萌芽は通常1つの切断された軸索に対して2〜5本でありビュングナー帯に入ってくることができる。この過程の結果、形態学的には再生するクラスターや再生繊維の薄い有髄線維の集まりとして観察される。近年ワーラー変性は軸索切断に至らない程度の軸索輸送のブロックでも生じることがわかり、dying-back型ニューロパチーとかなり共通したメカニズムと考えられている。
- dying-back型ニューロパチー
この軸索障害は、ある一群のニューロパチーに特徴的で、初めは軸索の最も遠位が障害され、ついで徐々により近位が変性する。ほぼ左右対称で亜急性もしくは慢性の変性を伴う。最も長く、大きい線維が初めに侵される。これを長さ依存性の脆弱性といい、多くの軸索性ニューロパチーで手袋靴下型で症状が出現する根拠となっている。脱髄型ニューロパチーでは長さ依存性の脆弱性は認められず、大腿部から症状が発現することもある。遠位部ほど神経細胞から栄養が届きにくいこと、あるいは神経毒素は軸索全般に作用するがそれに対する防御因子は遠位部ほど供給が少ないなどが機序として考えられている。病理学的所見は軸索の萌芽を含めた再生の証拠を伴う有髄線維の減少が特徴的である。後索の変性はこの機序では脊髄の上端から始まり末梢神経伝導速度検査では末期まで正常となることもある。
- ニューロノパチー
ニューロノパチーは細胞突起に属する軸索ではなく、神経細胞体の障害が最初であると考えられている軸索障害型ニューロパチーである。多少は細胞体障害を伴い、細胞体が障害されるため再生が不可能となっている。ニューロノパチーは、大脳皮質やその他の灰白質での神経細胞変性として、緩徐に進行し、引き続き選択的な神経細胞脱落を伴うことが特徴的である。ビタミンB6欠乏など中毒性ニューロノパチーや傍腫瘍性神経症候群では感覚神経の方が運動神経より障害されやすい。これは後根神経節では血液神経関門が欠いていることが関係すると考えられている。またファブリー病では小さな神経細胞が障害されやすく、傍腫瘍性神経症候群や感覚性ニューロパチーやフリードライヒ運動失調症、無βリポ蛋白血症では大きな神経細胞が障害されやすいといった神経細胞選択性も認められる。
原発性節性脱髄
原発性節性脱髄は一次性の髄鞘あるいはシュワン細胞の障害でおこる。運動軸索に起こった場合、軸索は正常であるため筋肉の脱神経による萎縮は起こりにくい。節性脱髄では線維の全長にわたって、髄鞘の節間が不連続になり、ある髄鞘が傷害される一方、その他は保たれる。病変は絞輪付近、paranodeからはじまる。シュワン細胞とマクロファージは変性した髄鞘を貪食する役割がある。ランビエ絞輪が広がったり、脱髄を起こした節間がむき出しになって伸びたりすることが、神経内の大部分の線維で起こると伝導ブロックをきたし、末梢神経伝導速度が低下する。脱髄が非常に狭い範囲で生じると(長さ15μm未満では)傷害をうけた節間に関与するシュワン細胞が髄鞘再形成を開始する。節間の消失が15μm以上では選択され、新しく増殖したシュワン細胞が髄鞘再形成を行い、小さく挿入された節間を形成する。この機序によってときほぐし像によるランビエ絞輪間距離のばらつきが生じる。節性脱髄と髄鞘再形成を繰り返すとオニオンバルブ(onion bulb)が形成される。脱髄後に軸索がむき出しになり、病態が収束すると軸索を失ったシュワン細胞や近傍のシュワン細胞などが増殖し始める。複数のシュワン細胞が1本の軸索をめぐって髄鞘化しようとし、基底膜内へと進入するが、成功するのは1つのシュワン細胞であり、他の細胞は周囲に追いやられる。ある細胞は死滅し、基底膜が残存する。この脱髄と再髄鞘化が繰り返し生じるとオニオンバルブを形成する。
病理診断としては再生繊維と節性脱髄の区別が難しい。有髄線維並の大きさの軸索がむき出しになり髄鞘がないときは脱髄が完成した線維と考える。一方薄いながら数層の髄鞘があるときは再生した有髄線維である。ときほぐし像でえられる脱随所見は脱髄が完成したものを見ているのであって、あれば確実な所見だが軽微な変化は捉えられない。
脱髄に至る過程は次の6種類に分類される。
- マクロファージによる髄鞘破壊
- ギラン・バレー症候群と慢性炎症性脱髄性ニューロパチーで特徴的に認められる。マクロファージがシュワン細胞の基底膜内に侵入し、髄鞘のintraperiod line(シュワン細胞の細胞膜外側どうしが癒合して生じた膜)を分離し分解していく。前段階として特異な抗体や補体の髄鞘への接着が想定される。
- vesicular demyelination(電子顕微鏡)
- 髄鞘が多数の小胞に分解される。剖検例で多く固定時のアーチファクトの可能性もあるが、糖尿病性ニューロパチーや薬物中毒などで多く認められる。
- widening of myelin lamella(電子顕微鏡)
- シュワン細胞膜の細胞外側どうしが癒着不全で離開するためintraperiod lineがみられない。間隙は細胞外の部分に広がる。小径線維に多く見られる傾向がある。IgMの単クローン性蛋白血症(κ鎖が多い)でMAG抗体を有する患者に特異的である。
- uncompacted myekin lamella(電子顕微鏡)
- 髄鞘のmajor dense lineで離開がありシュワン細胞の細胞質が髄鞘の中にみられる。POEMS症候群、遺伝性圧脆弱性ニューロパチー、CMT1Bなどでよくみられる。
- infolding and outfolding of myelin
- 髄鞘の部分的折りたたみ異常でおこる。ときほぐし像でトマキュラが認められる。遺伝性圧脆弱性ニューロパチー、CMT4B、CMT4F、CMT1A、抗MAG抗体陽性のニューロパチーで認められる。
- intramyelinic edema
- 髄鞘内の浮腫や空胞化である。慢性炎症性脱髄性ニューロパチーや抗MAG抗体陽性のニューロパチーに比較的特異的とされている。
特異所見と重要な所見
腓腹神経生検による診断において重要な所見をまとめる。
- 血管炎
- 血管炎では壊死性血管炎、微小血管炎、好酸球性肉芽腫が特異的な所見である。壊死性血管炎は顕微鏡的多発血管炎や好酸球性多発血管炎性肉芽腫症で見られることが多い。小血管で血管構築が破壊されており血管壁内の細胞浸潤が内膜、中膜、外膜の全層にわたってみられ、内幕と中膜を隔てる内弾性板が断裂しているとわかりやすい。微小血管炎はクリオグロブリン血症、非全身性血管炎性ニューロパチーで認められる。細動脈が炎症の場となるが毛管壁が薄いため血管構築の破壊を病理で証明するのは困難である。しばしばCD68抗体(マクロファージ)やCD3抗体(Tリンパ球)といった免疫染色を併用する。好酸球性肉芽腫は好酸球性多発血管炎性肉芽腫症で認められる。ステロイドが投与されると認められないことが多い。
- サルコイド肉芽腫
- 非乾酪性の肉芽腫で類上皮細胞、ラングハンス巨細胞、リンパ球からなる。神経よりも筋での陽性率の方が高い。肉芽腫のみならばサルコイドーシスのほかハンセン病、好酸球性多発血管炎性肉芽腫症、多発血管炎性肉芽腫症などでも認められる。
- アミロイド沈着
- 家族性アミロイドニューロパチーでは神経内鞘の血管周囲にアミロイド沈着が認められる。骨髄腫やALアミロイドーシスでも認められる。
- 活動性の脱髄
- マクロファージがとりついて髄鞘をはがし貪食している像やシュワン細胞が自壊して髄鞘が壊れる、すなわち軸索周囲に髄鞘崩壊産物が認められる場合は現在進行形の活動性脱髄である。
- 神経内鞘のリンパ球浸潤
- 1神経束に内鞘に2〜3個程度のリンパ球は病的ではないと考えられる。血管周囲を取り巻く多数のリンパ球浸潤はCIDPを示唆する。これをperivascular cuffingという。
- 規則的な髄鞘のほぐれ
- 電子顕微鏡所見であるがwidely spaced myelinは抗MAG抗体陽性のニューロパチー、Uncompacted myelin lamellaはPOEMS症候群で認められる。
- Onion bulb
- 有髄線維が脱髄と再髄鞘化を繰り返す過程でシュワン細胞が有髄線維を幾層にも玉ねぎ状にとりまいたものである。玉ねぎ状の取り巻きは通常4〜5層である。無髄線維や線維芽細胞を含むこともある。中心には髄鞘の薄い有髄線維があることも多いが軸索が残っていないこともある。CMT1A、CMT3、CIDPで認められる。
- Small onion bulb
- 軸索を取り巻く構造が1〜2層程度のものである。CMT1BやCIDP、MMN、IgMパラプロテインを伴うニューロパチー、糖尿病性ニューロパチー、Krabbe病などで認められる。
- Psedo-onion bulb
- 軸索変性後の再生線維はしばしばOnion bulbのようにみえる。中心の線維が2本以上あるいは不明瞭でとりまく成分に無髄線維を多数認める場合は再生繊維を疑う。
- 軸索腫大
- ノルマルヘキサンなどの中毒性ニューロパチーで認められる。軸索輸送障害で認められる。
- トマキュラ
- 局所的に髄鞘が過剰に取り巻き、厚さをましているものを指す。中心に軸索が残っているが軸索腫大はなくむしろ萎縮しているようにみえる。遺伝性圧脆弱性ニューロパチーで最も典型的に高頻度に認められる。その他はCMT4やIgMパラプロテインを伴うニューロパチーで認められる。
- 神経線維密度のばらつき
- 準特異的な所見である。後天性ニューロパチーを示唆する所見である。同じ神経束内でのばらつきの他に神経束どうしでばらつきが認められることもある。血管炎性ニューロパチー、サルコイドーシス、悪性リンパ腫などで認められる。近位部の病変で生検部位では軸索変性や軸索消失が認められるときもばらつきは出現する。
- 神経周膜の細胞浸潤
- 神経周膜の細胞浸潤はハンセン病、特発性神経周膜炎、クリオグロブリン血症、サルコイドーシス、ライム病で認められる。
- small fiber neuropathy
- 小径線維優位の脱落、変性はアミロイドニューロパチー(特に家族性の初期)、糖尿病性ニューロパチー(多発性感覚神経優位型)、急性自律性感覚性ニューロパチー、遺伝性感覚性ニューロパチー、ファブリー病、アルコール性ニューロパチーの一部、シェーグレン症候群の一部で認められる。
- 神経内鞘の浮腫
- 神経内鞘の浮腫は細胞成分がなく、トルイジンブルーで薄く染色される。神経周膜下に限局するものは病的意義はとぼしいが、内鞘の内部に広範囲に見られる場合は意義がある。POEMS症候群や多層性のonion bulbが認められる疾患CMT3、CIDP、Krabbe病などでよく認められる。
神経生検の標本の見方
まずは標本にアーチファクトの混入がないか確認する。次に神経束が何本あるのか確認する。2〜3本の場合は腓腹神経の分枝であり動脈も含まれていないことが多く、評価が不十分になる。有髄線維密度が保たれているか確認する。減っていたら何らかの軸索障害を意味する。大径線維、小径線維のそれぞれに選択性があるか確認する。異常所見の分布が均一か、局所的か評価する。局所的な場合は後天性疾患、特に虚血性、免疫性、感染性を示唆する。脱髄所見があるのか確認する。CIDPの診断支持基準ではときほぐし像で12%以上である。次に活動性の評価をする。ovoidや髄鞘の分解過程がないか確認する。これらは疾患の緊急度を示す。最後に特異的所見がないか確認する。
アトラス
 血管炎低倍率
血管炎低倍率 血管炎中倍率
血管炎中倍率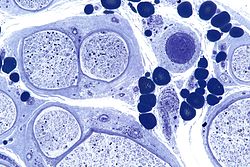 血管炎中倍率
血管炎中倍率 血管炎高倍率
血管炎高倍率 血管炎高倍率
血管炎高倍率 血管炎超高倍率
血管炎超高倍率






Remove ads
筋病理
→詳細は「筋病理学」を参照
脚注
参考文献
関連項目
外部リンク
Wikiwand - on
Seamless Wikipedia browsing. On steroids.
Remove ads