In many preparations of delicate organic compounds, specific parts of the molecules cannot survive the required reagents or chemical environments. These parts (functional groups) must be protected. For example, lithium aluminium hydride is a highly reactive reagent that usefully reduces esters to alcohols. It always reacts with carbonyl groups, and cannot be discouraged by any means. When an ester must be reduced in the presence of a carbonyl, hydride attack on the carbonyl must be prevented. One way to do so converts the carbonyl into an acetal, which does not react with hydrides. The acetal is then called a protecting group for the carbonyl. After the hydride step is complete, aqueous acid removes the acetal, restoring the carbonyl. This step is called deprotection.
As a rule, the introduction of a protecting group is straightforward. The difficulties honestly lie in their stability and in selective removal. Apparent problems in synthesis strategies with protecting groups are rarely documented in the academic literature.[2]
Orthogonal protection
Summarize
Perspective
Orthogonal protection of L-Tyrosine (Protecting groups are marked in blue, the amino acid is shown in black). (1) Fmoc-protected amino group, (2) benzyl ester protected carboxyl group and (3) tert-butyl ether protected phenolic hydroxyl group of Tyrosine.
Orthogonal protection is a strategy allowing the specific deprotection of one protective group in a multiply-protected structure. For example, the amino acid tyrosine could be protected as a benzyl ester on the carboxyl group, a fluorenylmethylenoxy carbamate on the amine group, and a tert-butyl ether on the phenol group. The benzyl ester can be removed by hydrogenolysis, the fluorenylmethylenoxy group (Fmoc) by bases (such as piperidine), and the phenolic tert-butyl ether cleaved with acids (e.g. with trifluoroacetic acid).
A common example for this application, the Fmoc peptide synthesis, in which peptides are grown in solution and on solid phase, is very important.[3] The protecting groups in solid-phase synthesis regarding the reaction conditions such as reaction time, temperature and reagents can be standardized so that they are carried out by a machine, while yields of well over 99% can be achieved. Otherwise, the separation of the resulting mixture of reaction products is virtually impossible (see also §Industrial applications).[4]
Schematic diagram of a solid-state peptide synthesis with orthogonal protecting groups X and Y
Fmoc solid state peptide synthesis with orthogonal protecting groups
A further important example of orthogonal protecting groups occurs in carbohydrate chemistry. As carbohydrates or hydroxyl groups exhibit very similar reactivities, a transformation that protects or deprotects a single hydroxy group must be possible for a successful synthesis.
Cleavage categorization
Many reaction conditions have been established that will cleave protecting groups. One can roughly distinguish between the following environments:[5]
Various groups are cleaved in acid or base conditions, but the others are more unusual.
Fluoride ions form very strong bonds to silicon; thus silicon protecting groups are almost invariably removed by fluoride ions. Each type of counterion, i.e. cleavage reagent, can also selectively cleave different silicon protecting groups depending on steric hindrance. The advantage of fluoride-labile protecting groups is that no other protecting group is attacked by the cleavage conditions.
Lipases and other enzymes cleave ethers at biological pH (5-9) and temperatures (30–40°C). Because enzymes have very high substrate specificity, the method is quite rare, but extremely attractive.
Catalytic hydrogenation removes a wide variety of benzyl groups: ethers, esters, urethanes, carbonates, etc.
Photolabile protecting groups bear a chromophore, which is activated through radiation with an appropriate wavelength and so can be removed.[6] For examples the o-nitrobenzylgroup ought be listed here.
The rare double-layer protecting group is a protected protecting group, which exemplify high stability.
Common protecting groups
Summarize
Perspective
Alcohol protecting groups
The classical protecting groups for alcohols are esters, deprotected by nucleophiles; triorganosilylethers, deprotected by acids and fluoride ions; and (hemi)acetals, deprotected by weak acids. In rarer cases, a carbon ether might be used.
The most important esters with common protecting-group use are the acetate, benzoate, and pivalate esters, for these exhibit differential removal. Sterically hindered esters are less susceptible to nucleophilic attack:
Chloroacetyl > acetyl > benzoyl > pivaloyl
Trimethylsilyl chloride, activated with imidazole, protects a secondary alcohol
Triorganosilyl sources have quite variable prices, and the most economical is chlorotrimethylsilane (TMS-Cl), a Direct Process byproduct. The trimethylsilyl ethers are also extremely sensitive to acid hydrolysis (for example silica gel suffices as a proton donator) and are consequently rarely used nowadays as protecting groups.
Aliphatic methyl ethers cleave with difficulty and only under drastic conditions, so that these are in general only used with quinonic phenols. However, hemiacetals and acetals are much easier to cleave.
Triethylsilyl— 10–100× stabler than a TMS group.[8] Cleaved with trifluoroacetic acid in water/tetrahydrofuran,[9] acetic acid in water/tetrahydrofuran,[10] or hydrogen fluoride in water or pyridine[11]
tert-Butyldimethylsilyl (TBDMS or TBS)— Cleaved with acetic acid in tetrahydrofuran/water,[12] Pyridinium tosylate in methanol,[13] trifluoroacetic acid in water,[14] hydrofluoric acid in acetonitrile,[15] pyridinium fluoride in tetrahydrofuran,[16]tetrabutylammonium fluoride in THF.[17] Commonly protects 2'-hydroxy function in oligonucleotide synthesis.
Triisopropylsilyl (TIPS) ethers)— Similar conditions to TBS but longer reaction times.[18]
tert‑Butyldiphenylsilyl (TBDPS)— Similar conditions to TBS but even longer reaction times (100–250× slower than TBS and 5–10× slower than TIPS)
Benzyl ethers:
Benzyl (Bn)— Removed by hydrogenolysis.[19] Bn group is widely used in sugar and nucleoside chemistry.
Trityl (triphenylmethyl, Tr)— Removed by acid[20][21][22] and hydrogenolysis
p,m‑Dimethoxybenzyl ether— Removed via oxidation with DDQ or ceric ammonium chloride[24]
Acetals:
Dimethoxytrityl, [bis-(4-methoxyphenyl)phenylmethyl] (DMT)— Removed by weak acid. DMT group is widely used for protection of 5'-hydroxy group in nucleosides, particularly in oligonucleotide synthesis.
Methoxytrityl [(4-methoxyphenyl)diphenylmethyl] (MMT) – Removed by acid and hydrogenolysis.
Benzyloxymethyl— Comparable stability to MOM, MEM und SEM,[25] but also admits reductive removal: sodium in liquid ammonia,[26][27] catalytic hydrogenation (palladium hydroxide on activated carbon), or Raney nickel in ethanol[28][29]
Ethoxyethyl ethers (EE) – Cleavage more trivial than simple ethers e.g. 1N hydrochloric acid[30]
Tris(isopropyl)silyloxymethyl (TOM)— Commonly protects 2'-hydroxy function in oligonucleotide synthesis.
β‑(Trimethylsilyl)ethoxymethyl— More labile than MEM and MOM to acid hydrolysis: 0.1M hydrochloric acid in methanol,[38] concentrated hydrofluoric acid in acetonitrile,[13] boron trifluoride etherate in dichloromethane,[39] or tetrabutylammonium fluoride in HMPT (Hexamethyl phosphoric acid triamide) or in tetrahydrofuran[40][41]
Other ethers:
p-Methoxyphenyl ether (PMP) – Removed by oxidation.[citation needed]
Allyl— Removed with potassium tert‑butoxide[43]DABCO in methanol, palladium on activated carbon, or diverse platinum complexes – conjoined with acid workup.[44]
Methyl ethers – Cleavage is by TMSI in dichloromethane or acetonitrile or chloroform. An alternative method to cleave methyl ethers is BBr3 in DCM
The 1,2‑diols (glycols) present for protecting-group chemistry a special class of alcohols. One can exploit the adjacency of two hydroxy groups, e.g. in sugars, in that one protects both hydroxy groups codependently as an acetal. Common in this situation are the benzylidene, isopropylidene and cyclohexylidene or cyclopentylidene acetals.
An exceptional case appears with the benzylideneprotecting group,which also admits reductive cleavage. This proceeds either through catalytic hydrogenation or with the hydride donor diisobutyl aluminum hydride (DIBAL). The cleavage with DIBAL deprotects one alcohol group, for the benzyl moiety stays as a benzyl ether on the second, sterically hindered hydroxy group.[45][46]
Amine protecting groups
BOC glycine. The tert-butyloxycarbonyl group is marked blue.
Amines have a special importance in peptide synthesis, but are a quite potent nucleophile and also relatively strong bases. These characteristics imply that new protecting groups for amines are always under development.[47]
Amine groups are primarily protected through acylation, typically as a carbamate. When a carbamate deprotects, it evolves carbon dioxide. The commonest-used carbamates are the tert-butoxycarbonyl, benzoxycarbonyl, fluorenylmethylenoxycarbonyl, and allyloxycarbonyl compounds.
Other, more exotic amine protectors are the phthalimides, which admit reductive cleavage,[48] and the trifluoroacetamides, which hydrolyze easily in base. Indoles, pyrroles und imidazoles— verily any aza-heterocycle— admit protection as N‑sulfonylamides,which are far too stable with aliphatic amines.[49]N‑benzylated amines can be removed through catalytic hydrogenation or Birch reduction, but have a decided drawback relative to the carbamates or amides: they retain a basic nitrogen.
Selection
Carbamates:
Carbamate group – Removed by acid and mild heating.
The most common protecting groups for carbonyls are acetals and typically cyclic acetals with diols. The runners-up used are also cyclic acetals with 1,2‑hydroxythiols or dithioglycols– the so-called O,S– or S,S-acetals.
Ethylene glycol1,3‑Propadiol
Overall, trans-acetalation plays a lesser role in forming protective acetals; they are formed as a rule from glycols through dehydration. Normally a simple glycol like ethylene glycol or 1,3-propadiol is used for acetalation.Modern variants also use glycols, but with the hydroxyl hydrogens replaced with a trimethylsilyl group.[60][61]
Acetals can be removed in acidic aqueous conditions. For those ends, the mineral acids are appropriate acids. Acetone is a common cosolvent, used to promote dissolution. For a non-acidic cleavage technique, a palladium(II) chloride acetonitrile complex in acetone[62] or iron(III) chloride on silica gel can be performed with workup in chloroform.[63]
Cyclic acetals are very much more stable against acid hydrolysis than acyclic acetals. Consequently acyclic acetals are used practically only when a very mild cleavage is required or when two different protected carbonyl groups must be differentiated in their liberation.[64]
Besides the O,O-acetals, the S,O- and S,S-acetals also have an application, albeit scant, as carbonyl protecting groups too. Thiols, which one begins with to form these acetals, have a very unpleasant stench and are poisonous, which severely limit their applications. Thioacetals and the mixed S,O-acetals are, unlike the pure O,O-acetals, very much stabler against acid hydrolysis. This enables the selective cleavage of the latter in the presence of sulfur-protected carbonyl groups.
The formation of S,S-acetals normally follows analogously to the O,O-acetals with acid catalysis from a dithiol and the carbonyl compound. Because of the greater stability of thioacetals, the equilibrium lies on the side of the acetal. In contradistinction to the O,O‑acetal case, it is not needed to remove water from the reaction mixture in order to shift the equilibrium.[65]
S,O-Acetals are hydrolyzed a factor of 10,000 times faster than the corresponding S,S-acetals. Their formation follows analogously from the thioalcohol. Also their cleavage proceeds under similar conditions and predominantly through mercury(II) compounds in wet acetonitrile.[66]
For aldehydes, a temporary protection of the carbonyl group the presence of ketones as hemiaminal ions is shown below. Here it is applied, that aldehydes are very much more activated carbonyls than ketones and that many addition reactions are reversible.[67][68]
Types of protectants
Acetals and Ketals – Removed by acid. Normally, the cleavage of acyclic acetals is easier than of cyclic acetals.
Dithianes – Removed by metal salts or oxidizing agents.
Carboxylic acid protecting groups
The most important protecting groups for carboxylic acids are the esters of various alcohols. Occasionally, esters are protected as ortho-esters or oxazolines.[69]
Many groups can suffice for the alcoholic component, and the specific cleaving conditions are contrariwise generally quite similar: each ester can be hydrolyzed in a basic water-alcohol solution. Instead, most ester protecting groups vary in how mildly they can be formed from the original acid.
Alkenes rarely need protection or are protected. They are as a rule only involved in undesired side reactions with electrophilic attack, isomerization or catalytic hydration. For alkenes two protecting groups are basically known:
Temporary halogenation with bromine to a trans‑1,2‑dibromoalkane: the regeneration of the alkene then follows with preservation of conformation via elemental zinc[86][87][88][89][90] or with titanocene dichloride.[91]
Protection through a Diels-Alder reaction: the transformation of an alkene with a diene leads to a cyclic alkene, which is nevertheless similarly endangered by electrophilic attack as the original alkene. The cleavage of a protecting diene proceeds thermically, for the Diels-Alder reaction is a reversible (equilibrium) reaction.[92][93][94]
Methyl (Me) – removed by strong nucleophiles e.c. thiophenole/TEA.
Terminal alkyne protecting groups
For alkynes there are in any case two types of protecting groups. For terminal alkynes it is sometimes important to mask the acidic hydrogen atom. This normally proceeds from deprotonation (via a strong base like methylmagnesium bromide or butyllithium in tetrahydrofuran/dimethylsulfoxide) and subsequently reaction with chlorotrimethylsilane to a terminally TMS-protected alkyne.[95] Cleavage follows hydrolytically– with potassium carbonate in methanol– or with fluoride ions like for example with tetrabutylammonium fluoride.[96]
Alkyne TMS protection
In order to protect the triple bond itself, sometimes a transition metal-alkyne complex with dicobalt octacarbonyl is used. The release of the cobalt then follows from oxidation.[97][98][99][100][101]
The use of protective groups is pervasive but not without criticism.[103] In practical terms their use adds two steps (protection-deprotection sequence) to a synthesis, either or both of which can dramatically lower chemical yield. Crucially, added complexity impedes the use of synthetic total synthesis in drug discovery. In contrast biomimetic synthesis does not employ protective groups. As an alternative, Baran presented a novel protective-group free synthesis of the compound hapalindole U. The previously published synthesis[104][105][106] according to Baran, contained 20 steps with multiple protective group manipulations (two confirmed):
Protected and unprotected syntheses of the marine alkaloid, hapalindole U.
Hideaki Muratake's 1990 synthesis using Tosyl protecting groups (shown in blue).
Phil Baran's protecting-group free synthesis, reported in 2007.
Industrial applications
Summarize
Perspective
Although the use of protecting groups is not preferred in industrial syntheses, they are still used in industrial contexts, e.g. sucralose (sweetener) or the Rochesynthesis of oseltamivir (Tamiflu, an antiviral drug)
An important example of industrial applications of protecting group theory is the synthesis of ascorbic acid (Vitamin C) à la Reichstein.
The Reichstein synthesis (of ascorbic acid)
In order to prevent oxidation of the secondary alcohols with potassium permanganate, they are protected via acetalation with acetone and then deprotected after the oxidation of the primary alcohols to carboxylic acids.[107]
A very spectacular example application of protecting groups from natural product synthesis is the 1994 total synthesis of palytoxin acid by Yoshito Kishi's research group.[108] Here 42 functional groups (39 hydroxyls, one diol, an amine group, and a carboxylic acid) required protection. These proceeded through 8 different protecting groups (a methyl ester, five acetals, 20 TBDMS esters, nine p‑methoxybenzyl ethers, four benzoates, a methyl hemiacetal, an acetone acetal and an SEM ester).[109]
Palytoxin
The introduction or modification of a protecting group occasionally influences the reactivity of the whole molecule. For example, diagrammed below is an excerpt of the synthesis of an analogue of Mitomycin C by Danishefsky.[110]
Part of the synthesis of an analogue of Mitomycin C with modified reactivity through protecting-group exchange
The exchange of a protecting group from a methyl ether to a MOM-ether inhibits here the opening of an epoxide to an aldehyde.
Protecting group chemistry finds itself an important application in the automated synthesis of peptides and nucleosides. The technique was introduced in the field of peptide synthesis by Robert Bruce Merrifield in 1977.[111] For peptide synthesis via automated machine, the orthogonality of the Fmoc group (basic cleavage), the tert‑butyl group (acidic cleavage) and diverse protecting groups for functional groups on the amino acid side-chains are used.[112] Up to four different protecting groups per nucleobase are used for the automated synthesis of DNA and RNA sequences in the oligonucleotide synthesis. The procedure begins actually with redox chemistry at the protected phosphorus atom. A tricoordinate phosphorus, used on account of the high reactivity, is tagged with a cyanoethyl protecting group on a free oxygen. After the coupling step follows an oxidation to phosphate, whereby the protecting group stays attached. Free OH-groups, which did not react in the coupling step, are acetylated in an intermediate step. These additionally-introduced protecting groups then inhibit, that these OH-groups might couple in the next cycle.[113]


 Schematic diagram of a solid-state peptide synthesis with orthogonal protecting groups X and Y
Schematic diagram of a solid-state peptide synthesis with orthogonal protecting groups X and Y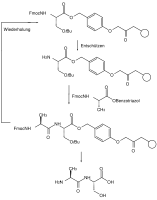 Fmoc solid state peptide synthesis with orthogonal protecting groups
Fmoc solid state peptide synthesis with orthogonal protecting groups





















