Loading AI tools

Vikipedi'den, özgür ansiklopediden
Progeria, halk dilindeki adıyla erken yaşlanma hastalığı, konu hakkında yapılan bilimsel araştırmalar hastalığın çaresini bulmaktan ziyade hastalığa sebep olan faktörleri bulmak ve bu sayede insanlığın ömrünü uzatabilmektir.
| Progeria | |
|---|---|
| Diğer adlar | Hutchinson-Gilford progeria sendromu (HGPS),[1][2] progeria sendromu,[2] Joseph Sendromu |
 | |
| Progeria hastası genç bir kız (solda). Sağlıklı bir hücre çekirdeği (sağda, üstte) ve progerik bir hücre çekirdeği (sağda, altta). | |
| Uzmanlık | Tıbbi genetik |
| Belirtiler | Büyüme gecikmesi, kısa boy, küçük yüz, saç dökülmesi |
| Komplikasyon | Kalp hastalığı, felç, kalça çıkığı[3] |
| Nedenleri | Genetik[3] |
| Tanı | Semptomlara dayalı olarak, genetik testler[3] |
| Ayırıcı tanı | Hallermann-Streiff sendromu, Gottron sendromu, Wiedemann-Rautenstrauch sendromu[3] |
| Tedavi | çoğunlukla semptomatik[3] |
| İlaç | Lonafarnib[4][5] |
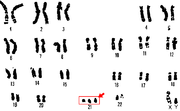
Diğer adı Hutchinson-Gilford olan bir genetik hastalık, ilk 46 kromozomla birlikte ortaya çıkan bu hastalık telomer zincirin kısalığından kaynaklanmaktadır. Normal somatik hücrelerde her replikasyon sonrası telomerik DNA kısalır. Bu DNA kısalmasını engellemek için vücutta telomeraz enzimi vardır. Telomeraz enzimi kromozomların kısalmasını engeller vücutta bu enzim eksikliği de bu hastalığa sebep olabilir. Telomer kısalması ve sınırlı yaşam süresi potansiyel tümör baskılama mekanizmasıdır. Çünkü insanın yapısındaki kromozomlar sürekli bölündüğü için kısalır bu da hücreleri yaşlandırır. Normalde ortalama 80 yıl yaşayan birey 12-13 yaşlarında yaşamını yitirir. Diğer bir deyişle henüz 10 yaşında 70 yaşındaki bir insan görünümüne sahiptir. 8 milyonda bir görülen bu hastalık için çalışmalar hastalığın sona ermesi yerine hastalıktaki kromozomların uzunluğunun dolayısıyla insanın yaşam süresinin uzatılması yönünde yapılmaktadır.
Hutchinson-Gilford Progeria Sendromu (HGPS) olarak da bilinen Progeria, öncelikle çocukları etkileyen nadir bir genetik bozukluktur. Bu durumun belirtileri tipik olarak yaşamın ilk birkaç ayı içinde ortaya çıkar. Erken belirtiler arasında gelişememe ve lokalize sklerodermaya benzeyen cilt değişiklikleri yer alabilir. Etkilenen çocuklar bebeklik dönemini geçtikçe, tipik olarak 18 ila 24 aylıkken ortaya çıkan ek ayırt edici özellikler belirgin hale gelir. Bu özellikler arasında sınırlı büyüme, tam saç dökülmesi (alopesi) ve girintili bir çene ve sıkışmış bir burun ile küçük bir yüz ile işaretlenmiş benzersiz bir yüz görünümü yer alır.
Çocuk yaşlanmaya devam ettikçe, progeria'nın ilerleyici doğası daha belirgin hale gelir. İlerlemiş semptomlar arasında kırışık cilt, böbrek yetmezliği, görme bozukluğu ve ateroskleroz gibi çeşitli kardiyovasküler sorunlar yer alabilir. Gövde ve ekstremitelerdeki derinin sertleşmesi ve sıkılaşması ile karakterize bir durum olan skleroderma, progeria hastalarında yaygın olarak görülür. Bu hastalık teşhisi konulan kişiler genellikle yaşlı yetişkinlere benzeyen küçük, kırılgan vücutlar sergilerler. Başları vücutlarına kıyasla nispeten büyüktür, dar, kırışık yüzleri ve gaga benzeri burunları vardır. Belirgin kafa derisi damarları, özellikle saç dökülmesi nedeniyle, belirgin gözlerle birlikte fark edilir hale gelir.
Kas-iskelet sistemi dejenerasyonu meydana gelerek vücut yağ ve kas kütlesinin kaybına, sert eklemlere, kalça çıkıklarına ve tipik olarak genç bireylerde görülmeyen diğer semptomlara yol açar. Bu fiziksel zorluklara rağmen, progeria hastası bireyler tipik zihinsel ve motor fonksiyonlarını sürdürürler.[6]
Hutchinson-Gilford sendromu (HGPS), yaşlanmayla ilişkilendirilen semptomların erken yaşta ortaya çıktığı son derece nadir bir otomal dominant genetik bozukluktur.[7] Genellikle sporadik gamet hattı mutasyonu sonucu ortaya çıkar; HGPS genetik olarak dominanttır, ancak insanlar genellikle bu bozukluğu kalıtsal olarak aktaracak kadar uzun yaşamazlar.[8]
.jpg)
Hücre çekirdeğinin yapısını zayıflatarak normal hücre bölünmesini zorlaştıran mutasyonlardan kaynaklanır. Bu mutasyonlar, lamin A adı verilen bir proteinin kodlanmasından sorumlu bir gende meydana gelir. Yapısal bir protein olan lamin A, son şekli olan lamin A'ya dönüşmek için bir dizi işlem adımından geçer. Ancak Progeria'lı bireylerde bu işlem bozulur ve anormal nükleer yapıya ve düzensiz kromatine yol açar.
Tipik durumlarda, LMNA geni, farnesilasyon ve metilasyon dahil olmak üzere çeşitli enzimatik modifikasyonları tetikleyen spesifik bir protein motifi (CAAX) içeren prelamin A'yı kodlar. Bu modifikasyonlar prelamin A'nın çekirdeğe girmesini sağlar ve burada lamin A'ya dönüşmek üzere parçalanır. Lamin A, diğer lamin proteinleri ile birlikte nükleer zarfın yapısal bütünlüğüne katkıda bulunur.
Progeria'da, LMNA genindeki spesifik bir mutasyon (pozisyon 1824), ekzon 11 içinde kriptik bir ekleme bölgesine neden olur. Bu da progerin adı verilen anormal bir protein varyantının üretilmesiyle sonuçlanır. Normal prelamin A'nın aksine, progerin bir bölünme bölgesinin olmaması nedeniyle farnesil grubunu çıkaramaz. Sonuç olarak, progerin nükleer kenara bağlı kalır ve nükleer zarfın yapısal desteğini tehlikeye atar. Bu yapısal zayıflık, hücre bölünmesi sırasında kromatin organizasyonunu bozarak hücrenin düzgün bölünme yeteneğini sınırlar.[9]
Ayrıca, progerin ifadesi DNA onarımını ve hücre polaritesinin oluşumunu da etkileyerek Progeria'nın gelişimine potansiyel olarak katkıda bulunur. Özellikle, Progeria'lı çocuklar doğumda normal görünürler ancak bir yaş civarında hastalık belirtileri göstermeye başlarlar.
LMNA geninde, mRNA, splicing veya protein amino asit dizisinde değişikliklere yol açabilen 1.400'den fazla Tek Nükleotid Polimorfizmi (SNP) tanımlanmıştır. Progerin üretimi, tipik yaşlanan hücrelerde aktive olduğu için normal yaşlanmada da rol oynayabilir.
Progeria'nın Werner sendromu, Cockayne sendromu veya xeroderma pigmentosum gibi yaşlanmanın belirli yönlerini etkileyen ancak hepsini birden etkilemeyen diğer hızlandırılmış yaşlanma hastalıklarından farklı olduğunu belirtmek gerekir. Bu hastalıklar genellikle "segmental progerias" olarak adlandırılır.
Progeria, yeni gebe kalmış bir zigotta veya bir ebeveynin gametlerinde hücre bölünmesi sırasında ortaya çıkan de novo dominant bir özellik olarak kabul edilir. Progeria Araştırma Vakfı'nı kuran Leslie Gordon ve Scott Berns, oğulları Sam'e genç yaşta bu hastalığın teşhisi konulduktan sonra Progeria konusunda farkındalık yaratılmasında ve araştırmaların ilerletilmesinde önemli bir rol oynamıştır.[10][11][12]