User:Benjah-bmm27/degree/4/SPT
From Wikipedia, the free encyclopedia
References
- Clayden, J. M. (2002). Organolithiums: Selectivity for Synthesis. Pergamon. ISBN 978-0080432625.
{{cite book}}: Unknown parameter|city=ignored (|location=suggested) (help) - Yamamoto, H.; Oshima, K., eds. (2004). Main Group Metals in Organic Synthesis. Wiley. ISBN 978-3527305087.
{{cite book}}: Unknown parameter|city=ignored (|location=suggested) (help) - Knochel, P.; Jones, P., eds. (1999). Organozinc Reagents: A Practical Approach. Oxford University Press. ISBN 978-0198501213.
- Taylor, R. J. K., ed. (1994). Organocopper Reagents: A Practical Approach. Oxford University Press. ISBN 978-0198557586.
- Krause, N., ed. (2002). Modern Organocopper Chemistry. Wiley. ISBN 978-3527297733.
{{cite book}}: Unknown parameter|city=ignored (|location=suggested) (help) - Carey, F. A.; Sundberg, R. J., eds. (2007). Advanced Organic Chemistry Part B: Reactions and Synthesis. Springer. ISBN 978-0387683546.
{{cite book}}: Unknown parameter|city=ignored (|location=suggested) (help)
Stereospecific and stereoselective reactions
General reminder:
- Stereospecific Reaction: A reaction in which the stereochemistry of the reactant completely determines the stereochemistry of the product without any other option.
- Stereoselective Reaction: A reaction in which there is a choice of pathway, but the product stereoisomer is formed due to its reaction pathway being more favourable than the others available.
Concise explanation from http://www.chem.ox.ac.uk/vrchemistry/nor/notes/stereo.htm
Insertion
In the insertion (reduction) method of preparing a main group organometallic reagent, a metallic main group element M reacts with an organohalide RX. The term reduction refers to the fact that the oxidation state of carbon in the organohalide decreases by two units. For example, MeCl → MeLi can be thought of as carbon(+1) → carbon(−1), or [H3C+ Cl−] → [H3C− Li+]. In reality, MeCl and MeLi are much more covalent than this ionic formulation, but it highlights the change in formal oxidation state.
- Halogen remains in the −1 oxidation state throughout
- Oxidation state of carbon decreases by two units
- Oxidation state of metal increases by two units (or two metals atoms are both oxidised by one unit): M → M2+ + 2e− or two lots of M → M+ + e−
- For a Group 1 metal: RX + 2M0 → RMI + MIX
- For a Group 2 metal: RX + M0 → RMIIX
The insertion reaction can be conducted on a large scale and is best for organobromides and organoiodides. Organochlorides usually require activation with zinc.
Metal-halogen exchange
- RX + R′M → RM + R′X
- Extremely fast - fast than deprotonation
- The reaction works if RM is less basic than R′M, i.e. the organometallic with the lowest pKaH is formed
- For example, BuLi + PhBr → PhLi + BuBr
- Consider PhI + tBuLi in Et2O and MeOH
- If deprotonation were faster than metal-halogen exchange, would observe route 1: tBuLi + MeOH → tBuH + LiOMe
- If metal-halogen exchange were faster than deprotonation, would observe route 2: tBuLi + PhI → tBuI + PhLi, then PhLi + MeOH → PhH + LiOMe
- PhH is the observed product, implying route 2 takes places, and metal-halogen exchange is faster than deprotonation
Transmetallation
In transmetallation, an organic group from an organometallic species is transferred to a different metal.
- Tin-lithium exchange is a common example
- R1SnBu3 + R2Li → Li+ [R1R2SnBu3]− → R1Li + R2SnBu3
- The best leaving group, R1, departs from [R1R2SnBu3]− as "R1−"
- RSnBu3 are bench-stable. Addition of BuLi generates Li[RSnBu4], which then decomposes to RLi + SnBu4. These products are easily separated by chromatography, so RSnBu3 are bench-stable stores of RLi.
- Example: PhSnBu3 + BuLi →→ SnBu4 + PhLi

Deprotonation
R-H + R′-M → R-M + R′-H
- Deprotonation of terminal alkynes by BuLi is common: R−C≡C-H + BuLi → R−C≡C-Li + BuH
- Requires the basicity of R′-M to be greater than that of R-M, i.e. R-H must be more acidic than R′-H
- pKaH BuLi, RMgX ~ 50
- pKaH R2N-M ~ 35
Organolithiums are common organic reagents. They are a source of "R−" and are very reactive towards electrophiles E+. They are often used to make other organometallic species by transmetallation.
Aggregation

- Organolithiums are oligomeric in solution - they form unreactive aggregates
- BuLi is a tetramer in solution: (BuLi)4
- tBuLi exists as a dimer in solution, (tBuLi)2 — this makes it easier to break up and thus more reactive
- Organolithium aggregates can be made more reactive by breaking them up with additives
Additives for disaggregating organolithium clusters
 Hexamethylphosphoramide (HMPA, most reactive but very toxic)
Hexamethylphosphoramide (HMPA, most reactive but very toxic) Pentamethyldiethylenetriamine (PMDTA)
Pentamethyldiethylenetriamine (PMDTA)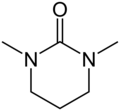 Dimethylpropyleneurea (DMPU, a safer alternative to HMPA)
Dimethylpropyleneurea (DMPU, a safer alternative to HMPA)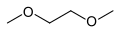 Dimethoxyethane (DME)
Dimethoxyethane (DME) Tetramethylethylenediamine (TMEDA)
Tetramethylethylenediamine (TMEDA)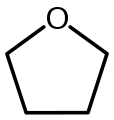 Tetrahydrofuran (THF)
Tetrahydrofuran (THF) Diethyl ether (Et2O, very slow to break aggregates)
Diethyl ether (Et2O, very slow to break aggregates)




- The additives are ligands that complete lithium's coordination sphere

Preparation
Insertion/Reduction
- R/Ar-Cl --[Li0]→ R/Ar-Li
- Works best with chlorides rather than bromides or iodides
- Rate of reaction is proportional to the stability of the radical R•
- The mechanism of reduction is single-electron transfer
Alkyl chlorides
- The rate-determining step (RDS) is the first step and involves a single electron from metallic lithium entering the C-Cl σ* orbital of tBuCl, breaking the C-Cl bond as a Cl-Li bond forms. The driving force for the reaction is the precipitation of insoluble LiCl.
- In the much faster second step, a tert-butyl radical tBu• combines with a neutral lithium atom Li• to form tBuLi

Aryl chlorides
- With aryl chlorides, the first step is reversible as the electron is entering a π* orbital
- Instead of concerted electron transfer and C-Cl bond fission as shown above, a radical anion intermediate is formed
- The radical anion slowly decomposes (RDS) to an aryl radical Ar• and LiCl
- Ar• and another Li• then combine to form the aryllithium ArLi

Arene-mediated reductive lithiation
- R/Ar-Cl + Li reactions don't work very well in practice, so an arene such as naphthalene is added as an electron shuttle
Naphthalene
- A lithium atom donates its valence electron to naphthalene, generating a radical anion
- The radical anion rapidly reduces the R/Ar-Cl to R/Ar•
- R/Ar• reacts with another lithium atom to form R/Ar-Li
- Problems: (i) R/Ar• can attack naphthalene, forming by-products and lowering yield, and (ii) naphthalene and its by-products can be difficult to separate from the desired product

DBB
- 4,4′-di-tert-butylbiphenyl (DBB) gives higher yields and is more recoverable than naphthalene
- Electron transfer can occur between species up to 7–9 Å, whereas bond formation requires less than 2 Å separation
- The bulky tert-butyl groups of DBB separate it enough from other molecules to avoid forming bonds (and thus by-products), but allow sufficiently close approach for electron transfer

Lithium-halogen exchange
Mechanism of transmetallation
Tin-lithium exchange
Deprotonation
Superbases
Enantioselective deprotonations
Reaction of organolithiums with electrophiles
Carbonyls
Orbital considerations
Lithiated carbamates
Rearrangements
Shapiro reaction
Bamford–Stevens reaction
Brook rearrangement
Wittig rearrangements
- 1,2-Wittig rearrangement
- 1,4-Wittig rearrangement
Grignard reagents
- Discovered by Victor Grignard in 1900, for which he won the 1912 Nobel Prize in Chemistry
- They have a more covalent metal-carbon bond than organolithiums, and are less pyrophoric
- A wide range of Grignard reagents are commercially available
Schlenk equilibrium
In ether solution, dissociation of Grignard reagents occurs:
2 R–Mg–X ⇌ R–Mg–R + X–Mg–X
Organomagnesium iodides, RMgI, exist primarily as R–Mg–R in THF.
References
- Silverman, G. S.; Rakita, P. E. (1996). Handbook of Grignard Reagents. Marcel Dekker. ISBN 978-0824795450.
{{cite book}}: Unknown parameter|city=ignored (|location=suggested) (help) - Richey, Jr., H. G., ed. (1999). Grignard Reagents: New Developments. Wiley. ISBN 978-0471999089.
{{cite book}}: Unknown parameter|city=ignored (|location=suggested) (help)
Preparation of Grignard reagents
Insertion/reduction
R–X + Mg0 ⇌ R–Mg–X
Groups that react with Grignard reagents inhibit Grignard formation completely
- At the temperature required for Grignard reagents to form (warm enough for Mg to insert into C–X bond), the newly formed C–Mg group will react with the following functional groups:
- Aldehydes RCHO and ketones RCOR
- Nitriles RCN
- Nitro compounds, RNO2
- Esters RCO2R, carboxylic acids RCO2H, amides RCONR2
- A leaving groups (such as tosylate) beta to MgX will be expelled, forming an alkene
Mechanism of Grignard reagent formation
- Single electron transfer, as for organolithiums (see above)
Transmetallation and magnesium-halogen exchange
- Although standard Grignard formation does not occur well below 0 °C, magnesium-halogen exchange is rapid
- At these low temperatures, Grignard reagents do not react with many functional groups, including esters
- They do still react with aldehydes and ketones, however
- It is therefore possible to prepare Grignards bearing ester groups (which would react with themselves at higher temperatures) by Mg-X exchange
- The usual reagent is iPrMgCl, which has bulky isopropyl groups
- It is added to aryl bromides or chlorides at, say, −20 °C or −35 °C
- The arylmagnesium halide formed by magnesium-halogen exchange
- It can then react with an aldehyde or ketone
Knochel reactions in synthesis
Reactions of Grignards with electrophiles
Carbonyls
Differences in reactivity between RLi and RMgX
Overview
1,4-Addition of RCu to enones
Reaction of RCu with RX
Carbocupration
1,4-Addition
- Corey, E. J.; Boaz, N. W. (1985). Tet. Lett. 26: 6015–6018. doi:10.1016/S0040-4039(00)95113-X.
{{cite journal}}: Missing or empty|title=(help)
- Reaction of R2CuLi with certain chiral enones leads to 92:8 of one diastereomer (the thermodynamic product)
- Adding Me3SiCl to the reaction mixture gives > 99:1 of the other diastereomer (the kinetic product)
- These results suggest the cuprate addition is reversible unless trimethylsilyl chloride is present to trap the enolate intermediate
- The key step is oxidative addition of the C=C bond of the enone to CuI, forming CuIII
- J. Am. Chem. Soc. 129: 7208–7209. 2008. doi:10.1021/ja067533d.
{{cite journal}}: Missing or empty|title=(help)
- J. Am. Chem. Soc. 129: 7208–7209. 2008. doi:10.1021/ja067533d.
Mechanisms
Kinetics
- 1,4-addition is first order in (Me2CuLi)2 – two equivalents of Me2CuLi
- Proceeds somewhat like a Grignard reaction
- The rate determining step is reductive elimination of the enolate product from the CuIII intermediate
- Krauss, S. R.; Smith, S. G. (1981). J. Am. Chem. Soc. 103: 141–148. doi:10.1021/ja00391a026.
{{cite journal}}: Missing or empty|title=(help)
R2CuLi cluster
- Readable account: Carey and Sundberg, Part B, chapter 8
- Hardcore account: Nakamura; et al. (1997). J. Am. Chem. Soc. 119: 4900–4910. doi:10.1021/ja964209h.
{{cite journal}}: Explicit use of et al. in:|author=(help); Missing or empty|title=(help)- All sorts of complicated equilibria and intermediate structures
- A nightmare to remember for the exam! Are we really expected to memorise this?
Asymmetric 1,4-addition
Conjugate reduction of enones

Stryker's reagent
- Need a soft source of H− to favour addition at the 4 position
- Ph3P + CuCl --[1. tBuONa, 2. H2]→ [(Ph3P)CuH]6 — Stryker's reagent, a red crystalline solid, 50-65 %
- React with enone in benzene at room temperature for 28 h
- Acts as "H-Cu", irreversible addition of hydride, under kinetic control
Asymmetric enone reduction
- Use (S)-p-tol-BINAP instead of Ph3P as a ligand for Cu, [{(S)-p-tol-BINAP}CuH]
- Use polymethylhydrosiloxane (PMHS), (SiHMeO)n, as a very stable source of hydride
- React with enone in toluene at room temperature for 22 h
- Can even tolerate aldehydes — selective 1,4-addition, c.f. NaBH4/LiBH4
Overview
- Organozinc reagents are highly tolerant of functional groups - the least reactive R-M
- Undergo facile transmetallation
- Highly reactive with H2O and O2
- Need a Lewis base (LB) to activate organozincs — they're unreactive when linear but reactive when bent by coordination of an LB
Addition to enones
Addition to aldehydes
Asymmetric addition to aldehydes
Simmons-Smith reaction
- The Simmons-Smith reaction is the conversion of an alkene to a cyclopropane by reaction with CH2I2 and Zn/Cu
- (Z)-alkenes give cis-cyclopropanes, (E)-alkenes give trans-cyclopropanes
Mechanism
- Syn addition of CH2 to the alkene
- Zn inserts into a C-I bond, forming I-Zn-CH2I, which acts like the carbene :CH2, being both electrophilic and nucleophilic at C
- Five-centred transition state
- Two C-Zn bonds form, C=C, C-I and C-Zn bonds break
Alcohol-directed Simmons-Smith
- Cyclic allylic and homoallylic alcohols have an OH group fixed above one side of the C=C bond
- This OH group coordinates to Zn in IZnCH2I, directing addition of CH2 to the same face of the alkene
- If the OH group is further away than homoallylic, no directing effect is observed and a racemic mixture of products is formed
Asymmetric Simmons-Smith
- Developed by André Charette at the Université de Montréal
- Requires allylic alcohols
- Uses a cyclic boronic ester as a stoichiometric chiral ligand: B-Zn transmetallation?
- Deployed in the synthesis of the natural product V-106305, which contains five trans cyclopropanes in a row
Hydroboration
Asymmetric hydroboration
- Enantioselective syn addition of R2B–H across C=C of an alkene
- Diisopinocampheylborane (Ipc2BH) + Z/cis-alkene → Ipc2B–alkyl, 87% ee
Rhodium-catalysed hydroboration
- The topic of GCLJ's PhD with J. Brown. Hyashi also investigated.
- R–CH=CH2 + HB(OR)2 (catecholborane) --[Rh(I)Ln]→ R–CH2–CH2–B(OR)2 (β) or R–CHMe–B(OR)2 (α)
- RhI catalysts tend to give the branched α-product (Markovnikov addition)
- Catalytic cycle involes four major steps:
- Oxidative addition of H–B to Rh(I)Ln
- Coordination of the alkene to Rh(III)
- Hydride transfer to the alkene (hydrorhodation) – becomes alkyl–Rh (selectivity-determining step)
- Reductive elimination of the boronic ester RB(OR)2
Oxidation of organoboranes
To alcohols
- H2O2 and NaOH convert R3B to ROH
- Retention of B–C stereochemistry due to orbital requirements of the mechanism
- Mechanism:
- HOO− and R3B form an ate-complex [R3B–OOH]−
- A 1,2-metallate rearrangement (stereospecific, antiperiplanar step) sees an R-group migrate from B to O, expelling OH− in the process
- A boronic ester R2B–O–R is the product
- This is hydrolysed to the alcohol ROH by NaOH/H2O
To amines
- H2N–OSO3H converts R3B to RNH2
- Mechanism:
- H2N–OSO3H and R3B form an ate-complex [R3B–NH–OSO3H]−
- An R-group migrates from B to N, expelling OSO3H−, leaving R–NH–BR2
- R–NH–BR2 is hydrolysed to RNH2 by H2O
Carbonyl reduction
- RCO2H is reduced to RCH2OH by BH3
- Very selective for carboxylic acids, even in the presence of aldehydes, ketones (which are more reactive), amides and esters
- Mechanism:
- The OH oxygen of RCO2H forms an ate complex with BH3, losing H+ to give RC(=O)–O–BH2
- H–BH2 then adds across C=O, forming R–CH(OBH2)2
- Some further (not given in lectures) step(s) occur to give the alcohol
1,2-Metallate rearrangement
- H. Brown, D. Matteson, D. Hoppe. P. Kocienski, VKA
- Addition of "R−" from R–M to (RO)2B–CR′2(LG) gives ate-complex [(RO)2BR–CR′2(LG)]−
- R migrates from B to C, expelling LG in the process, generating RR′2C–B(OR)2 (the actual 1,2-metallate rearrangement step)
- RR′2C–B(OR)2 can be oxidised to RR′2C–OH
- The 1,2-metallate rearrangement step is stereospecific, requiring antiperiplanar R–B and C-LG bonds and involving inversion at carbon
Matteson
- Add LiCHCl2 to R′–B(OR)2, where (OR)2 is actually a chiral bidentate "ligand" for B
- Initial ate-complex formed is [R′–B(OR)2–CHCl2]−
- Undergoes 1,2-met to R′–CHCl–B(OR)2 with loss of Cl−
- Add a Grignard R″–MgX to R′–CHCl–B(OR)2, attack at B is faster than SN2 at C–Cl σ*, forming [R′–CHCl–B(OR)2–R″]−
- Another 1,2-met: R″ migrates from B to C, expelling the second chloride, undergoing inversion at C, and forming R′R″HC–B(OR)2
- R′R″HC–B(OR)2 is then oxidised to R′R″HC–OH or R′R″HC–NH2
- Two inversions at carbon lead to overall retention at carbon, stereospecific reaction
VKA: lithiation-borylation
- Enantioselective deprotonation (s-BuLi, (−)-sparteine) converts a carbamate to a lithiated carbamate
- The lithiated carbamate forms an ate-complex with a pinacol-boronic ester RBpin
- 1,2-met: R migrates from B to C, expelling OCb, forming a different pinacol-boronic ester
Wikiwand in your browser!
Seamless Wikipedia browsing. On steroids.
Every time you click a link to Wikipedia, Wiktionary or Wikiquote in your browser's search results, it will show the modern Wikiwand interface.
Wikiwand extension is a five stars, simple, with minimum permission required to keep your browsing private, safe and transparent.