Protein found in humans From Wikipedia, the free encyclopedia
Succinate dehydrogenase complex subunit C, also known as succinate dehydrogenase cytochrome b560 subunit, mitochondrial, is a protein that in humans is encoded by the SDHCgene.[5] This gene encodes one of four nuclear-encoded subunits that comprise succinate dehydrogenase, also known as mitochondrial complex II, a key enzyme complex of the tricarboxylic acid cycle and aerobic respiratory chains of mitochondria. The encoded protein is one of two integral membrane proteins that anchor other subunits of the complex, which form the catalytic core, to the inner mitochondrial membrane. There are several related pseudogenes for this gene on different chromosomes. Mutations in this gene have been associated with pheochromocytomas and paragangliomas. Alternatively spliced transcript variants have been described.[6]
The gene that codes for the SDHC protein is nuclear, even though the protein is located in the inner membrane of the mitochondria. The location of the gene in humans is on the first chromosome at q21. The gene is partitioned in six exons. The SDHC gene produces an 18.6kDa protein composed of 169 amino acids.[7][8]
The SDHC protein is one of the two transmembrane subunits of the four-subunit succinate dehydrogenase (Complex II) protein complex that resides in the inner mitochondrial membrane. The other transmembrane subunit is SDHD. The SDHC/SDHD dimer is connected to the SDHB electron transport subunit which, in turn, is connected to the SDHA subunit.[9]
The SDHC protein is one of four nuclear-encoded subunits that comprise succinate dehydrogenase, also known as Complex II of the electron transport chain, a key enzyme complex of the citric acid cycle and aerobic respiratory chains of mitochondria. The encoded protein is one of two integral membrane proteins that anchor other subunits of the complex, which form the catalytic core, to the inner mitochondrial membrane.[6]
SDHC forms part of the transmembrane protein dimer with SDHD that anchors Complex II to the inner mitochondrial membrane. The SDHC/SDHD dimer provides binding sites for ubiquinone and water during electron transport at Complex II. Initially, SDHA oxidizes succinate via deprotonation at the FAD binding site, forming FADH2 and leaving fumarate, loosely bound to the active site, free to exit the protein. The electrons derived from succinate tunnel along the [Fe-S] relay in the SDHB subunit until they reach the [3Fe-4S] iron sulfur cluster. The electrons are then transferred to an awaiting ubiquinone molecule at the Q pool active site in the SDHC/SDHD dimer. The O1 carbonyl oxygen of ubiquinone is oriented at the active site (image 4) by hydrogen bond interactions with Tyr83 of SDHD. The presence of electrons in the [3Fe-4S] iron sulphur cluster induces the movement of ubiquinone into a second orientation. This facilitates a second hydrogen bond interaction between the O4 carbonyl group of ubiquinone and Ser27 of SDHC. Following the first single electron reduction step, a semiquinone radical species is formed. The second electron arrives from the [3Fe-4S] cluster to provide full reduction of the ubiquinone to ubiquinol.[10]
Mutations in this gene have been associated with paragangliomas.[6][11] More than 30 mutations in the SDHC gene have been found to increase the risk of hereditary paraganglioma-pheochromocytoma type 3. People with this condition have paragangliomas, pheochromocytomas, or both. An inherited SDHC gene mutation predisposes an individual to the condition, and a somatic mutation that deletes the normal copy of the SDHC gene is needed to cause hereditary paraganglioma-pheochromocytoma type 3. Most of the inherited SDHC gene mutations change single amino acids in the SDHC protein sequence or result in a shortened protein. As a result, there is little or no SDH enzyme activity. Because the mutated SDH enzyme cannot convert succinate to fumarate, succinate accumulates in the cell. The excess succinate abnormally stabilizes hypoxia-inducible factors (HIF), which also builds up in cells. Excess HIF stimulates cells to divide and triggers the production of blood vessels when they are not needed. Rapid and uncontrolled cell division, along with the formation of new blood vessels, can lead to the development of tumors in people with hereditary paraganglioma-pheochromocytoma.[12]
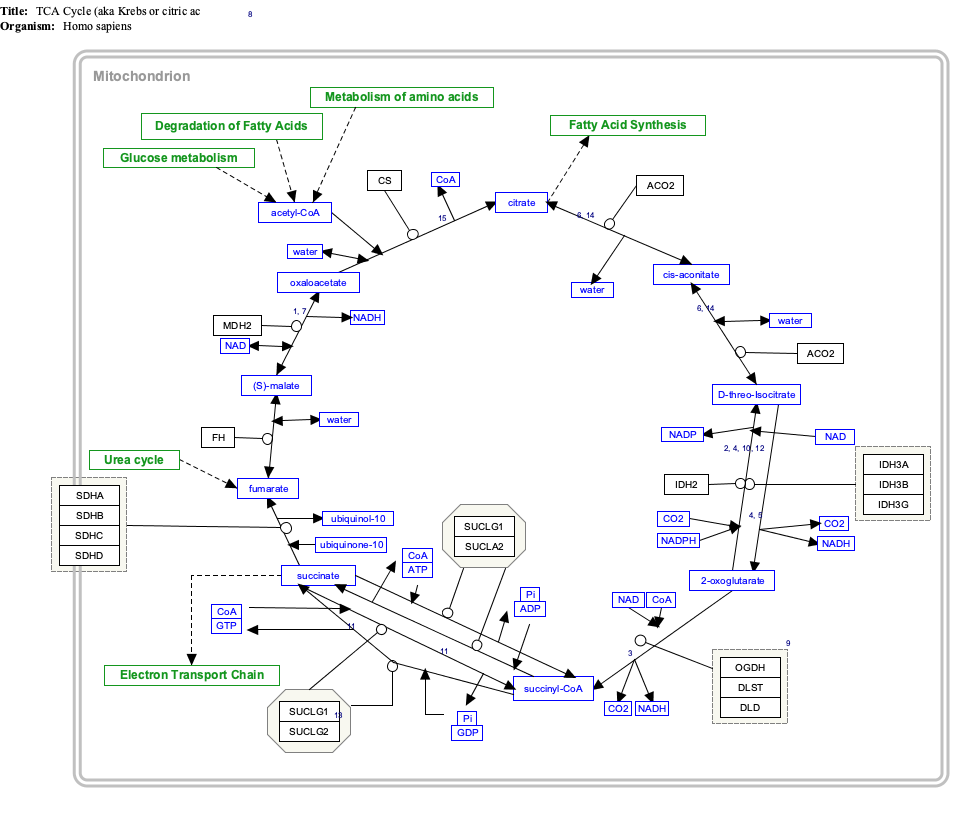
Click on genes, proteins and metabolites below to link to respective articles.[§ 1]
Hirawake H, Taniwaki M, Tamura A, Kojima S, Kita K (1997). "Cytochrome b in human complex II (succinate-ubiquinone oxidoreductase): cDNA cloning of the components in liver mitochondria and chromosome assignment of the genes for the large (SDHC) and small (SDHD) subunits to 1q21 and 11q23". Cytogenetics and Cell Genetics. 79 (1–2): 132–8. doi:10.1159/000134700. PMID9533030.
Niemann S, Müller U, Engelhardt D, Lohse P (July 2003). "Autosomal dominant malignant and catecholamine-producing paraganglioma caused by a splice donor site mutation in SDHC". Human Genetics. 113 (1): 92–4. doi:10.1007/s00439-003-0938-0. PMID12658451. S2CID32412131.
Cascón A, López-Jiménez E, Landa I, Leskelä S, Leandro-García LJ, Maliszewska A, Letón R, de la Vega L, García-Barcina MJ, Sanabria C, Alvarez-Escolá C, Rodríguez-Antona C, Robledo M (September 2009). "Rationalization of genetic testing in patients with apparently sporadic pheochromocytoma/paraganglioma". Hormone and Metabolic Research. 41 (9): 672–5. doi:10.1055/s-0029-1202814. PMID19343621. S2CID24979281.
Goto Y, Ando T, Naito M, Goto H, Hamajima N (2006). "No association of an SDHC gene polymorphism with gastric cancer". Asian Pacific Journal of Cancer Prevention. 7 (4): 525–8. PMID17250422.
Cascón A, Pita G, Burnichon N, Landa I, López-Jiménez E, Montero-Conde C, Leskelä S, Leandro-García LJ, Letón R, Rodríguez-Antona C, Díaz JA, López-Vidriero E, González-Neira A, Velasco A, Matias-Guiu X, Gimenez-Roqueplo AP, Robledo M (May 2009). "Genetics of pheochromocytoma and paraganglioma in Spanish patients". The Journal of Clinical Endocrinology and Metabolism. 94 (5): 1701–5. doi:10.1210/jc.2008-2756. PMID19258401.
Boedeker CC, Neumann HP, Maier W, Bausch B, Schipper J, Ridder GJ (July 2007). "Malignant head and neck paragangliomas in SDHB mutation carriers". Otolaryngology–Head and Neck Surgery. 137 (1): 126–9. doi:10.1016/j.otohns.2007.01.015. PMID17599579. S2CID13650583.
Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY, Richardson AL, Sidhu SB, Robinson BG, Clifton-Bligh RJ (June 2010). "Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes". Human Pathology. 41 (6): 805–14. doi:10.1016/j.humpath.2009.12.005. PMID20236688.
Eng C, Kiuru M, Fernandez MJ, Aaltonen LA (March 2003). "A role for mitochondrial enzymes in inherited neoplasia and beyond". Nature Reviews. Cancer. 3 (3): 193–202. doi:10.1038/nrc1013. PMID12612654. S2CID20549458.
Richalet JP, Gimenez-Roqueplo AP, Peyrard S, Vénisse A, Marelle L, Burnichon N, Bouzamondo A, Jeunemaitre X, Azizi M, Elghozi JL (December 2009). "A role for succinate dehydrogenase genes in low chemoresponsiveness to hypoxia?". Clinical Autonomic Research. 19 (6): 335–42. doi:10.1007/s10286-009-0028-z. PMID19768395. S2CID2265162.
Pigny P, Cardot-Bauters C, Do Cao C, Vantyghem MC, Carnaille B, Pattou F, Caron P, Wemeau JL, Porchet N (February 2009). "Should genetic testing be performed in each patient with sporadic pheochromocytoma at presentation?". European Journal of Endocrinology. 160 (2): 227–31. doi:10.1530/EJE-08-0574. PMID19029228. S2CID31746044.
Korpershoek E, Van Nederveen FH, Dannenberg H, Petri BJ, Komminoth P, Perren A, Lenders JW, Verhofstad AA, De Herder WW, De Krijger RR, Dinjens WN (August 2006). "Genetic analyses of apparently sporadic pheochromocytomas: the Rotterdam experience". Annals of the New York Academy of Sciences. 1073 (1): 138–48. Bibcode:2006NYASA1073..138K. doi:10.1196/annals.1353.014. PMID17102080. S2CID8418586.