Carbon–hydrogen bond activation
Organic reactions in which the H in a C–H bond is substituted From Wikipedia, the free encyclopedia

In organic chemistry and organometallic chemistry, carbon–hydrogen bond activation (C−H activation) is a type of organic reaction in which a carbon–hydrogen bond is cleaved and replaced with a C−X bond (X ≠ H is typically a main group element, like carbon, oxygen, or nitrogen). Some authors further restrict the term C–H activation to reactions in which a C–H bond, one that is typically considered to be "unreactive", interacts with a transition metal center M, resulting in its cleavage and the generation of an organometallic species with an M–C bond. The organometallic intermediate resulting from this step (sometimes known as the C−H activation step) could then undergo subsequent reactions with other reagents, either in situ (often allowing the transition metal to be used in a catalytic amount) or in a separate step, to produce the functionalized product.[1]
The alternative term C−H functionalization is used to describe any reaction that converts a relatively inert C−H bond into a C−X bond, irrespective of the reaction mechanism (or with an agnostic attitude towards it). In particular, this definition does not require the cleaved C–H bond to initially interact with the transition metal or for an organometallic intermediate to exist in the reaction mechanism.[2] In contrast to the organometallic variety, this broadened type of C-H activation is widely employed industrially and in nature. This broader definition encompasses all reactions that would fall under the restricted definition of C–H activation given above. However, it also includes iron-catalyzed alkane C–H hydroxylation reactions that proceed through the oxygen rebound mechanism (e.g. cytochrome P450 enzymes and their synthetic analogues), in which an organometallic species is not believed to be involved in the mechanism. In other cases, organometallic species are indirectly involved. This occurs, for example, with Rh(II)-catalyzed C–H insertion processes in which an electrophilic metal carbene species is generated and the hydrocarbon C–H bond inserts into the carbene carbon without direct interaction of the hydrocarbon with the metal. Other mechanistic possibilities not involving direct C–H bond cleavage by the metal include (i) generation of arylmetal species by electrophilic aromatic substitution mechanism (common for electrophilic Pd, Pt, Au, Hg species), (ii) cleavage of the C–H bond via hydrogen atom abstraction by an O- or N-centered radical, which may then go on to further react and undergo functionalization with or without forming an organometallic intermediate (e.g., Kharasch–Sosnovsky reaction), and (iii) C–H deprotonation at the α-position of a π-system assisted by initial formation of a π-complex with an electrophilic metal to generate a nucleophilic organometallic species (e.g., by cyclopentadienyliron complexes).
Often, when authors make the distinction between C–H functionalization and C−H activation, they will restrict the latter to the narrow sense. However, it may be challenging to definitively demonstrate the involvement or non-involvement of an interaction between the C–H bond and the metal prior to cleavage of the bond. This article discusses C–H functionalization reactions in general but with a focus on C–H activation sensu stricto.
Classification
Mechanisms for C-H activation by metal centers can be classified into three general categories:
- (i) Oxidative addition, in which a low-valent metal center inserts into a carbon-hydrogen bond, which cleaves the bond and oxidizes the metal:
- LnM + RH → LnM(R)(H)
- (ii) Electrophilic activation in which an electrophilic metal attacks the hydrocarbon, displacing a proton:
- LnM+ + RH → LnMR + H+
- One particularly commonly variant of this category, known as concerted metalation–deprotonation, involves a ligated internal base (often a carboxylate, e.g., acetate or pivalate) simultaneously accepting the displaced proton intramolecularly.
- (iii) Sigma-bond metathesis, which proceeds through a "four-centered" transition state in which bonds break and form in a single step:
- LnMX + RH → LnMR + XH
Historic overview
Summarize
Perspective
The first C–H activation reaction is often attributed to Otto Dimroth, who in 1902, reported that benzene reacted with mercury(II) acetate (See: organomercury). Many electrophilic metal centers undergo this Friedel-Crafts-like reaction. Joseph Chatt observed the addition of C-H bonds of naphthalene by Ru(0) complexes.[3]
Chelation-assisted C-H activations are prevalent. Shunsuke Murahashi reported a cobalt-catalyzed chelation-assisted C-H functionalization of 2-phenylisoindolin-1-one from (E)-N,1-diphenylmethanimine.[4]

In 1969, A.E. Shilov reported that potassium tetrachloroplatinate induced isotope scrambling between methane and heavy water. The pathway was proposed to involve binding of methane to Pt(II). In 1972, the Shilov group was able to produce methanol and methyl chloride in a similar reaction involving a stoichiometric amount of potassium tetrachloroplatinate, catalytic potassium hexachloroplatinate, methane and water. Due to the fact that Shilov worked and published in the Soviet Union during the Cold War era, his work was largely ignored by Western scientists. This so-called Shilov system is today one of the few true catalytic systems for alkane functionalizations.[1][5]
In some cases, discoveries in C-H activation were being made in conjunction with those of cross coupling. In 1969,[6] Yuzo Fujiwara reported the synthesis of (E)-1,2-diphenylethene from benzene and styrene with Pd(OAc)2 and Cu(OAc)2, a procedure very similar to that of cross coupling. On the category of oxidative addition, M. L. H. Green in 1970 reported on the photochemical insertion of tungsten (as a Cp2WH2 complex) in a benzene C–H bond[7] and George M. Whitesides in 1979 was the first to carry out an intramolecular aliphatic C–H activation[8]

The next breakthrough was reported independently by two research groups in 1982. R. G. Bergman reported the first transition metal-mediated intermolecular C–H activation of unactivated and completely saturated hydrocarbons by oxidative addition. Using a photochemical approach, photolysis of Cp*Ir(PMe3)H2, where Cp* is a pentamethylcyclopentadienyl ligand, led to the coordinatively unsaturated species Cp*Ir(PMe3) which reacted via oxidative addition with cyclohexane and neopentane to form the corresponding hydridoalkyl complexes, Cp*Ir(PMe3)HR, where R = cyclohexyl and neopentyl, respectively.[9] W.A.G. Graham found that the same hydrocarbons react with Cp*Ir(CO)2 upon irradiation to afford the related alkylhydrido complexes Cp*Ir(CO)HR, where R = cyclohexyl and neopentyl, respectively.[10] In the latter example, the reaction is presumed to proceed via the oxidative addition of alkane to a 16-electron iridium(I) intermediate, Cp*Ir(CO), formed by irradiation of Cp*Ir(CO)2.
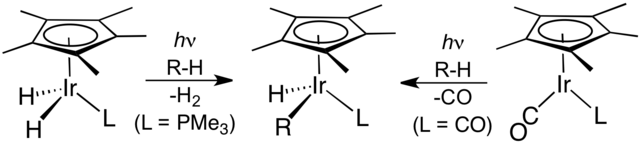
C–H activation by Bergman et al. (left) and Graham et al.

The selective activation and functionalization of alkane C–H bonds was reported using a tungsten complex outfitted with pentamethylcyclopentadienyl, nitrosyl, allyl and neopentyl ligands, Cp*W(NO)(η3-allyl)(CH2CMe3).[11]

C–H activation of pentane, as seen in Ledgzdins et al., J. Am. Chem. Soc. 2007; 129, 5372–3.

In one example involving this system, the alkane pentane is selectively converted to the halocarbon 1-iodopentane. This transformation was achieved via the thermolysis of Cp*W(NO)(η3-allyl)(CH2CMe3) in pentane at room temperature, resulting in elimination of neopentane by a pseudo-first-order process, generating an undetectable electronically and sterically unsaturated 16-electron intermediate that is coordinated by an η2-butadiene ligand. Subsequent intermolecular activation of a pentane solvent molecule then yields an 18-electron complex possessing an n-pentyl ligand. In a separate step, reaction with iodine at −60 °C liberates 1-iodopentane from the complex.
Mechanistic understanding
Summarize
Perspective
One approach to improving chemical reactions is the understanding of the underlying reaction mechanism. time-resolved spectroscopic techniques can be used to follow the dynamics of the chemical reaction. This technique requires a trigger for initiating the process, which is in most cases illumination of the compound. Photoinitiated reactions of transition metal complexes with alkanes serve as a powerful model systems for understanding the cleavage of the strong C-H bond.[9][10]

In such systems, the sample is illuminated with UV-light, which excites the metal center, leading to ligand dissociation. This dissociation creates a highly reactive, electron deficient 16-electron intermediate, with a vacant coordination site. This species then binds to an alkane molecule, forming a σ-complex (coordination of a C-H bond). In a third step, the metal atom inserts into the C-H bond, cleaving it and yielding the alkyl (or aryl) metal hydride.
The intermediates and their kinetics can be observed using time-resolved spectroscopic techniques (e.g. TR-IR, TR-XAS, TR-RIXS). Time-resolved infrared spectroscopy (TR-IR) is a rather convenient method to observe these intermediates. However, it is only limited to complexes which have IR-active ligands and is prone to correct assignments on the femtosecond timescale due to underlying vibrational cooling. To answer the question of difference in reactivity for distinct complexes, the electronic structure of those needs to be investigated. This can be achieved by X-ray absorption spectroscopy (XAS) or resonant inelastic X-ray scattering (RIXS). These methods have been used to follow the steps of C-H activation with orbital resolution and provide detailed insights into the responsible interactions for the C-H bond breaking.[12][13]
Full characterization of the structure of methane bound to a metal center was reported by Girolami in 2023: isotopic perturbation of equilibrium (IPE) studies involving deuterated isotopologs showed that methane binds to the metal center through a single M···H-C bridge; changes in the 1JCH coupling constants indicate clearly that the structure of the methane ligand is significantly perturbed relative to the free molecule.[14]
Directed C-H activation
Summarize
Perspective
Directed-, chelation-assisted-, or "guided" C-H activation involves directing groups that influence regio- and stereochemistry.[15] This is the most useful style of C-H activation in organic synthesis. N,N-dimethylbenzylamine undergoes cyclometalation readily by many transition metals.[16] A semi-practical implementations involve weakly coordinating directing groups, as illustrated by the Murai reaction.[17]

The mechanism for the Pd-catalyzed C-H activation reactions of 2-phenylpyridine involves a metallacycle intermediate. The intermediate is oxidized to form a PdIV species, followed by reductive elimination to form the C-O bond and release the product.[18]

Borylation
Transforming C-H bonds into C-B bonds through borylation has been thoroughly investigated due to their utility in synthesis (i.e. for cross-coupling reactions). John F. Hartwig reported a highly regioselective arene and alkane borylation catalyzed by a rhodium complex. In the case of alkanes, exclusive terminal functionalization was observed.[19]

Later, ruthenium catalysts were discovered to have higher activity and functional group compatibility.[20]

Other borylation catalysts have also been developed, including iridium-based catalysts, which activate C-H bonds with high compatibility.[21][22][23]
For more information, consult borylation.
Natural gas
Although chemists have failed to develop a commercial process for selective C-H activation of methane, such a reaction is the basis of reverse methanogenesis. In this nickel-catalyzed process, methane is converted to the methyl substituent of coenzyme M, CH3SCH2CH2SO−3.[24]
Naturally occurring methane is not utilized as a chemical feedstock, despite its abundance and low cost. Current technology makes prodigious use of methane by steam reforming to produce syngas, a mixture of carbon monoxide and hydrogen. This syngas is then used in Fischer-Tropsch reactions to make longer carbon chain products or methanol, one of the most important industrial chemical feedstocks.[25][26] An intriguing method to convert these hydrocarbons involves C-H activation. Roy A. Periana, for example, reported that complexes containing late transition metals, such as Pt, Pd, Au, and Hg, react with methane (CH4) in H2SO4 to yield methyl bisulfate.[27][28] The process has not however been implemented commercially.

Asymmetric C–H functionalization

Rhodium-catalyzed carbene insertion reactions have been rendered enantioselective using chiral carboxylate ligands, notably the proline-derived DOSP ligand.[30]
The total synthesis of lithospermic acid employs guided C–H functionalization late stage to a highly functionalized system. The directing group, a chiral nonracemic imine, is capable of performing an intramolecular alkylation, which allows for the rhodium-catalyzed conversion of imine to the dihydrobenzofuran.[31]

Enantioselective C–H silylation has emerged as a useful strategy for introducing well-defined stereochemistry at silicon or at nearby carbon centers.[32][33]
Applications toward the synthesis of natural products and bioactive molecules
The total synthesis of calothrixin A and B features an intramolecular Pd-catalyzed cross coupling reaction via C-H activation, an example of a guided C-H activation. Cross coupling occurs between aryl C-I and C-H bonds to form a C-C bond.[34] The synthesis of a mescaline analogue employs the rhodium-catalyzed enantioselective annulation of an aryl imine via a C-H activation.[35]

Alkene isomerization
One type of useful transition metal C-H bond activations are alkene isomerization. At least two mechanisms are recognized. For alkene-metal hydrides, isomerization can proceed via migratory insertion, followed by beta-hydride elimination. This process is the basis of chain walking. Another mechanism for alkene isomerization is the conversion of an alkene complex to an allyl-hydride complex.[36]
See also
Older reviews
- Pre-2004
- Arndtsen, B. A.; Bergman, R. G.; Mobley, T. A.; Peterson, T. H. (1995). "Selective Intermolecular Carbon–Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution". Accounts of Chemical Research. 28 (3): 154–162. doi:10.1021/ar00051a009.
- Crabtree, R. H. (2001). "Alkane C–H activation and functionalization with homogeneous transition metal catalysts: a century of progress – a new millennium in prospect". J. Chem. Soc., Dalton Trans. 17 (17): 2437–2450. doi:10.1039/B103147N.
- 2004-7
- Crabtree, R. H. (2004). "Organometallic alkane CH activation". J. Organomet. Chem. 689 (24): 4083–4091. doi:10.1016/j.jorganchem.2004.07.034. S2CID 95482372.
- Organometallic C–H Bond Activation: An Introduction Alan S. Goldman and Karen I. Goldberg ACS Symposium Series 885, Activation and Functionalization of C–H Bonds, 2004, 1–43
- Periana, R. A.; Bhalla, G.; Tenn, W. J.; III; Young, K. J. H.; Liu, X. Y.; Mironov, O.; Jones, C.; Ziatdinov, V. R. (2004). "Perspectives on some challenges and approaches for developing the next generation of selective, low temperature, oxidation catalysts for alkane hydroxylation based on the C–H activation reaction". Journal of Molecular Catalysis A: Chemical. 220 (1): 7–25. doi:10.1016/j.molcata.2004.05.036.
- Lersch, M.Tilset (2005). "Mechanistic Aspects of C−H Activation by Pt Complexes". Chem. Rev. 105 (6): 2471–2526. doi:10.1021/cr030710y. PMID 15941220., Vedernikov, A. N. (2007). "Recent Advances in the Platinum-mediated CH Bond Functionalization". Curr. Org. Chem. 11 (16): 1401–1416. doi:10.2174/138527207782418708.
- 2008-2011
- Davies, H. M. L.; Manning, J. R. (2008). "Catalytic C–H functionalization by metalcarbenoid and nitrenoid insertion". Nature. 451 (7177): 417–424. Bibcode:2008Natur.451..417D. doi:10.1038/nature06485. PMC 3033428. PMID 18216847.
- Boutadla, Y.; Davies, D. L.; Macgregor, S. A.; Poblador-Bahamonde, A. I. (2009). "Mechanisms of C–H bond activation: rich synergy between computation and experiment". Dalton Trans. 2009 (30): 5820–5831. doi:10.1039/B904967C. PMID 19623381.
- Wencel-Delord, J.; Dröge, T.; Liu, F.; Glorius, F. (2011). "Towards Mild Metal-Catalyzed C–H Bond Activation". Chem. Soc. Rev. 40 (9): 4740–4761. doi:10.1039/C1CS15083A. PMID 21666903.
- Shulpin, G. B. (2010). "Selectivity enhancement in functionalization of C–H bonds: A review". Org. Biomol. Chem. 8 (19): 4217–4228. doi:10.1039/c004223d. PMID 20593075.
- Lyons, T. W.; Sanford, M. S. (2010). "Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions". Chem. Rev. 110 (2): 1147–1169. doi:10.1021/cr900184e. PMC 2836499. PMID 20078038.*Balcells, D.; Clot, E.; Eisenstein, O. (2010). "C–H Bond Activation in Transition Metal Species from a Computational Perspective". Chem. Rev. 110 (2): 749–823. doi:10.1021/cr900315k. PMID 20067255.
- 2012-2015
- Hashiguchi, B. G.; Bischof, S. M.; Konnick, M. M.; Periana, R. A. (2012). "Designing Catalysts for Functionalization of Unactivated C–H Bonds Based on the CH Activation Reaction". Acc. Chem. Res. 45 (6): 885–898. doi:10.1021/ar200250r. PMID 22482496.
- Kuhl, N.; Hopkinson, M. N.; Wencel-Delord, J.; Glorius, F. (2012). "Beyond Directing Groups: Transition Metal-Catalyzed C H Activation of Simple Arenes". Angew. Chem. Int. Ed. 51 (41): 10236–10254. doi:10.1002/anie.201203269. PMID 22996679.
- Wencel-Delord, J.; Glorius, F. (2013). "C–H bond activation enables the rapid construction and late-stage diversification of functional molecules". Nature Chemistry. 5 (5): 369–375. Bibcode:2013NatCh...5..369W. doi:10.1038/nchem.1607. PMID 23609086.
Additional sources
References
Wikiwand - on
Seamless Wikipedia browsing. On steroids.